
In-Depth Report
Welcome to ERS 2021

Anita Simonds, President of the ERS, welcomed delegates to the second virtual ERS congress at the end of another remarkable year. Respiratory teams have been challenged by the demands of the pandemic while continuing to manage the needs of their acute and chronic respiratory patients. The main aims of ERS this year have been to advance its digital health agenda, make its voice heard on health policy issues and celebrate respiratory multi-disciplinary teams.
Holistic care in ILD: monitoring the patient’s pathway
In this session to explore the lessons learnt from home monitoring of patients during COVID-19, Ganesh Raghu, Seattle, USA, began the presentations by considering the application of telemedicine to evaluate patients with interstitial lung disease (ILD).
The use of telemedicine has increased dramatically over the past decade in the field of respiratory medicine with some experts concluding that it can be as effective as traditional face-to-face consultations. During the pandemic, the need for continued care of chronic health conditions forced physicians and healthcare systems to adopt a hybrid model, offering telehealth in addition to usual care in-person visits. Telemedicine offers considerable benefits to the patient, such as minimal interruptions to their schedule, access to experts from any location and potential to participate in clinical trials. It is cost effective and minimises risk of exposure to potential infection in clinic.
“Is telemedicine an unintended consequence of COVID-19.... a sliver lining?”
Ganesh Raghu, Seattle, USA
However, there are challenges in using telemedicine in the broad field of ILD, which represents a heterogeneous group of complex acute and chronic disease. Firstly, evaluation may be less than optimal. Secondly, there is the risk that lack of direct contact in three-dimensional space interferes with cues on a subconscious level and, thirdly, it does not build trust and confidence with patients and their families. Ultimately, the advantage of convenience from telemedicine has to be weighed against the benefits of in-person consultations.

Home monitoring of ILD was explored further by Marlies Wijsendbeek, Rotterdam, The Netherlands. Almost 300 patients with ILD were monitored at home during the pandemic and 98% have opted to continue using the app. Patient experiences relating to home monitoring were positive, with more medication adjustments, no increase in anxiety or depression and good adherence. Home spirometry is feasible and reliable for regular care and is also suitable for safety monitoring in the research setting. However, it should not be relied upon as a primary endpoint because the number of measures impacts on the ability to run statistical testing. It shows promise as a tool to predict disease progression but further studies are required to investigate this.
Elisabeth Bendstrup, Aarhus, Denmark, discussed palliative care in ILD. Breathlessness is a major debilitating symptom for these patients and management should involve optimal treatment of comorbidities, non-pharmacological and pharmacological approaches. Practical strategies for living well have been described by patients with idiopathic pulmonary fibrosis (IPF), such as physically slowing down, taking one stair at a time, and maintaining a positive mental attitude. Breathing techniques like singing and humming can be helpful as well as avoiding extreme temperatures and humidity.

Four practical strategies for living well, as described by patients with IPF
It is important that patients and caregivers receive practical assistance with end-of-life planning. A post-hoc analysis in non-malignant patients has shown that advance care planning leads to significantly longer survival.
In the final presentation of the session, Anne-Marie Russell, London, UK, talked about the journey towards patient-centred care that has taken place over the last fifty years. Both healthcare professionals (HCPs) and patients are now acutely aware of the importance of understanding the whole person and adopting a biopsychosocial (as opposed to biomedical) perspective. This is achieved with management through a multi-disciplinary team and communication throughout the interdisciplinary team.

MDT coordination of care
“Nowadays, patients recognise that disease may be biological in origin but their impact is felt physically, psychologically and socially.”
Anne-Marie Russell, London, UK
Post-COVID ILD syndrome: diagnosis and management
In this debate, two speakers were tasked with making cases for or against the use of antifibrotic drugs in patients with post-COVID ILD syndrome.
COVID-19 pneumonia is a severe profibrotic condition. Results from a study conducted in Wuhan, China, demonstrated persistent functional alterations 12 months after acute COVID-19 pneumonia, particularly in those who experienced severe illness. Bruno Crestani, Paris, France, argued that this long-term lung damage is due to persistent fibrotic alterations. Abnormalities commonly include traction bronchiectasis, honeycombing and parenchymal bands with altered lung function and dyspnoea.
Dr Crestani proposed a strong rationale for using antifibrotic agents in the treatment of persistent fibrosis of COVID-19. IPF and COVID-19 ILD syndrome share common risk factors and pathophysiology and these are the same pathophysiological pathways that are targeted by current antifibrotics. An initial study on the use of nintendanib in COVID-19 patients reported no effect on survival but it was promising that treatment reduced the time spent on ventilation. There are currently four active trials listed on Clinicaltrials.gov so more data will be forthcoming (NCT04607928; NCT04282902; NCT04653831; NCT04856111).

Risk factors and pathophysiological features common to IPF and COVID-19
“There is a strong rationale for antifibrotics in late COVID-ILD”
Bruno Crestani, Paris, France
On the other side of the debate, Sara Tomassetti, Florence, Italy, argued that the case for first-line use of antifibrotics is not clear. She described an early auto-inflammatory phenotype of COVID-19 ILD syndrome that has been shown to respond to corticosteroid treatment. Dr Tomassetti argued that the majority of cases have an underlying auto-inflammatory pathogenic background and only a minority are fibrotic cases with potential links to IPF. According to the interim analysis of her team’s Post COvid Interstitial Lung Syndrome (PCOILS) trial, only 18% of 550 high-resolution computed tomography (HRCT) images from post-COVID ILD patients at 6 months showed diffuse fibrotic changes in >5% of total lung. Eleven percent were classified as non-specific interstitial pneumonia (NSIP), or organising pneumonia (OP) for which there is no indication to treat with antifibrotic drugs first-line but only in the case of disease progression despite immunomodulation. The remaining 7% were either definite/ probable or indeterminate for usual interstitial pneumonia (UIP) for which there is no indication to treat with antifibrotics unless cases are clinically reclassified as IPF or progressive fibrotic ILDs. She suggested that there are two main fibrotic phenotypes: incidental diagnosis IPF and ‘true’ post-COVID fibrosis. Dr Tomassetti concluded, therefore, that immunomodulation should be first-line treatment in most of these patients while antifibrotics should be considered only in selected cases.
“Immunomodulation seems currently the most appropriate first-line treatment in the majority of post-COVID ILD cases”
Sara Tomassetti, Florence, Italy
Imaging grand round
Lucio Calandriello, Rome, Italy, considered the use of imaging in IPF. The diagnostic approach to IPF is highly reliant on images of the lungs, but diagnosis can be challenging since over 50% of cases of UIP have atypical chest HRCT appearances. Most cases of familial interstitial pneumonia do not conform to typical UIP pattern on HRCT despite evidence of UIP pattern at the histologic level. It is also challenging that there is a discrepancy in advice between the American Thoracic Society (ATS) and the Fleischner Society on when a biopsy is not warranted. In cases where IPF is suspected, the ATS recommends that biopsy would not be required with typical UIP, however, the Fleischner Society White Paper recommends that biopsy would not be required in cases of either typical or probable UIP.

Recommendations from society guidelines on whether a biopsy is required for diagnosis of IPF
Simon Walsh, London, UK, considered the multi-disciplinary diagnosis from the perspective of the radiologist. There are a few specific diagnoses that can be made with 100% confidence based on the HRCT alone, for example, dendriform ossification. However, the more common conditions can only be made based on a broad knowledge of the patient’s history and other results so a multi-disciplinary approach is required. In over half of cases, radiologists change their initial diagnosis once they have access to additional information gleaned during a multi-disciplinary meeting (MDM), in particular smoking history, disease behaviour, the presence of underlying connective tissue disease, lung function data and bronchoalveolar lavage results. Age and gender are also vital to the diagnosis but these would be included on the HRCT.
State of the art session: ILDs
Coline Van Moorsel, Nieuwegein, The Netherlands, discussed familial IPF, which is commonly defined as being present in two or more first-degree relatives. It is a progressive disease with a median survival of 2–5 years so it is important to recognise it early in order to optimise treatment. Multiple phenotypes have been reported and pathogenic mutation has been identified in about 40% of cases (N=221). The majority of these mutations relate to short telomeres, which are associated with more severe disease and earlier onset.
Telomere length is heritable and shortens with every generation of mutation carriers, meaning that age of disease onset is similar among siblings and earlier in offspring. Since family members are at increased risk for pulmonary fibrosis (PF) or short telomere syndrome, should we be routinely screening first-degree relatives of patients? The ERS Task Force on Genetics in Pulmonary Fibrosis will publish a statement next year providing guidance on genetic testing in this field.

Statement on genetic pulmonary fibrosis expected 2022
“Studying familial disease provides an opportunity to understand the natural history of PF and in this way we might discover markers for preclinical disease and develop targeted treatment”
Coline Van Moorsel, Nieuwegein, The Netherlands
Andreas Guenther, Giessen, Germany, talked about ILD registries and what we can learn from them. There are currently many multicentre registries in the US, Europe, Asia and Australia and there is ongoing debate about the creation of a global registry, a development which Dr Guenther believes would be highly valuable.
International, multicentre registries in IPF/ILD serve as valuable tools to confirm that results from clinical trials can be replicated under real-world conditions. They can also be used to analyse regional or national differences in clinical approaches to management of IPF/ILD and answer important questions relating to comorbidities, treatments and risk factors. The combination of registry data with state-of-the-art biobanking is even more valuable as they can be used to identify diagnostic, prognostic and therapeutic biomarkers, and conduct experiments in ex vivo human models. Unfortunately, few registries currently offer biobanking activities.
One group that does offer both registry and biobank is the European IPF network. This database captures a vast amount of clinical data on over 3000 parameters. Researchers can apply to have access to biomaterials for translational research, biomarker development and development of novel treatment modalities. Many publications resulting from clinical and observational studies have come from data in this registry, and over 40 publications from biomaterials alone.
“I think the biggest advantages can be gained when you combine registries with biobank data”
Andreas Guenther, Giessen, Germany
Martin Kolb, Ontario, Canada, talked about progressive fibrosing ILDs. There are over 200 types of ILD, with IPF making up less than a quarter of all cases. In 1285 patients with ILD in the Canadian Registry for PF, a third of cases were due to connective tissue disease (CTD-ILD) while another 22% are unclassifiable. During the course of disease, some non-IPF ILDs will develop progressive fibrosis leading to increased symptoms, decreased quality of life and early mortality. It is important to recognise that antifibrotic drugs target progressive fibrosis and are not specific for IPF and non-IPF ILDs can benefit from treatment. There are several randomised, controlled trials for antifibrotics in systemic sclerosis ILD (SSc-ILD). Phase II data suggest that antifibrotics may improve lung function in patients with non-IPF progressive ILDs, such as rheumatoid arthritis ILD (RA-ILD), chronic hypersensitivity pneumonitis, or unclassifiable interstitial pneumonia, and further controlled studies are warranted.
In the final presentation, Abdellatif Tazi, Paris, France, talked about pulmonary Langerhans cell histiocytosis (PLCH). It is a rare ILD of unknown aetiology, characterised by organ/tissue infiltration by Langerhans-like dendritic cells. PLCH lesions destroy the walls of bronchioles showing a destructive bronchiolitis pattern on HRCT along with reduced lung function. Presentation can be a single system disease, most commonly bone, or multisystem in two organs or more (including lung). It is a disease that occurs almost exclusively in smokers. Recent studies have identified several activation mutations in the mitogen-activated protein kinase (MAPK) pathway in patients with PLCH.
Lungs on fire: ILD?
 This session was a series of four real-life clinical cases which Nazia Chaudhuri, Manchester, UK, presented to a panel of experts who were encountering these cases for the first time. Members of the expert panel were Katerina Antoniou, Greece, and Carlos Robalo Cordeiro and Tiago Alfaro, both from Portugal.
This session was a series of four real-life clinical cases which Nazia Chaudhuri, Manchester, UK, presented to a panel of experts who were encountering these cases for the first time. Members of the expert panel were Katerina Antoniou, Greece, and Carlos Robalo Cordeiro and Tiago Alfaro, both from Portugal.
Case 1 Contributed by Mário Alexandere Oliveira Pinto, Portugal
Sjogren’s syndrome Systemic AA amyloidosis with renal, salivary gland and pleural involvement
- Pertinent points in case
- 73-year-old female
- History of arterial hypertension, chronic kidney disease and chronic musculoskeletal pain
- Referred to nephrologist and presented with dyspnoea
- Thoracentesis found mononucleate cells in exudative pleural fluid, which was sterile and negative for malignant cells
- Meanwhile, renal biopsy showed amyloid deposits in the interstitium and vascular walls, staining positive for AA
- Further questioning revealed xerostomia (dry mouth) and Raynaud’s phenomenon, which combined with musculoskeletal pain, suggested autoimmune disease
- Autoimmune panel screening was positive for ANA, anti-Ro/SSA and anti-La/SSB antibodies
- Salivary gland biopsy showed chronic grade III sialadenitis and amyloid deposits
- Treatment and outcome
- Corticosteroid (prednisolone) and steroid-sparing agent (azathioprine) to counter pleural effusion
- Renal function deteriorated and multiple infectious complications led to death several months later
- Key learnings
- AA amyloidosis is caused by tissue deposition of serum AA protein and is associated with prolonged inflammatory states. Treatment is directed at the underlying condition
- Pleural effusions can occur in systemic amyloidosis, and one third are exudative
- Pleural infiltration by amyloid is rare, occurring in 1–2% of systemic amyloidosis
- Pleural effusions of amyloid origin are refractory to diuretics, relapse quickly after thoracentesis and confer poor prognosis
AA, A-amyloid; ANA, antinuclear antibodies
Case 2 Contributed by Sophie Cottin, Belgium
Anti-TNF induced ILD secondary to adalimumab
- Pertinent points in case
- 61-year-old male
- History of chronic psoriasis, type 2 diabetes and hepatic steatosis
- Adalimumab treatment initiated 15 months previously for psoriasis
- Presented with shortness of breath, mild haemoptysis and bi-basal crackles
- Laboratory results showed a very low inflammatory syndrome and non-specific ANA and ANCA
- Treatment and outcome
- Adalimumab treatment discontinued
- Thoracic HRCT scans at 1, 2 and 5 months showed reversal of lung damage
- Key learnings
- Anti-TNF-induced ILD is now recognised as a specific entity given the expansion of use of biological therapy
- Different morphological patterns have been described, including NSIP and diffuse alveolar damage
- Pathological mechanisms are unclear
- Treatment discontinuation is recommended and should not be reintroduced or replaced by another drug belonging to the same class. Adjunctive corticosteroids can be considered.
ANA, antinuclear antibodies; ANCA, anti-neutrophil cytoplasmic antibodies; HRCT, high-resolution computed tomography;
ILD, interstitial lung disease; NSIP, non-specific interstitial pneumonia; TNF, tumour necrosis factor
Case 3 Contributed by Johad Khoury, Israel
IgG4-related disease
- Pertinent points in case
- 63-year-old male, non-smoker
- Worsening dyspnoea over 4 years and non-productive cough
- Lung function tests showed reduced diffusion capacity
- Unremarkable blood results and echocardiography
- Chest HRCT showed enlarged lymph node and oesophageal varices
- Hepatology consultation identified cryptogenic liver cirrhosis
- Lung and liver biopsies were not performed due to risk of complications
- IgG4-related disease was considered because of PF, enlarged lymph node and cryptogenic liver disease
- Lymph node plasma showed serum IgG levels were elevated
- Treatment and outcome
- Adalimumab treatment discontinued
- Thoracic HRCT scans at 1, 2 and 5 months showed reversal of lung damage
- Key learnings
- IgG4-related disease is a rare immune-mediated fibro-inflammatory condition that is capable of affecting multiple organs, including inflammatory hepatitis and interstitial pneumonitis
- Affected lung tissue exhibits characteristic lymphoplasmacytic infiltrates enriched in IgG4-positive plasma cells, usually with abundant fibrosis
- Four patterns of lung involvement have been described: solid nodular, bronchovascular, round-shaped GG opacities, and alveolar interstitial (with honeycombing, bronchiectasis and GG opacities)
GG, ground glass; HRCT, high-resolution computed tomography; IgG, immunoglobulin G; PF, pulmonary fibrosis
Case 4 Contributed by Wallace Wee, Canada
Eosinophilic granulomatosis with polyangiitis
- Pertinent points in case
- 10-year-old child, referred for asthma management
- Asthma from 8 years of age and recurrent pneumonias every 4–6 weeks from age 7
- History of dysphagia and poor weight gain
- Presents with worsening dysphagia, recurrent pneumonias, non-specific abdominal discomfort and poor weight gain
- Blood tests show elevated eosinophils
- Abdominal ultrasound showed thickened loops of bowel
- Endoscopy showed eosinophilic esophagitis and chronic gastritis
- MRI of gastrointestinal tract showed small bowel and rectal wall thickening, lymphoma and eosinophilic enteropathy
- Bone marrow biopsy showed eosinophilia of all stages of maturation
- Diagnosed with eosinophilic granulomatosis with polyangiitis based on asthma, eosinophilia, non-fixed pulmonary infiltrates and extravascular eosinophils
- Treatment and outcome
- Treated with mepolizumab and continued with inhaled corticosteroids. Systemic steroids were considered
- Key learnings
- ANCA is detected in 30–35% of eosinophilic granulomatosis with polyangiitis patients
- Glucocorticoids are the mainstay therapy
- Mepolizumab may be useful as a steroid-sparing agent and likely needed as lifelong treatment to maintain remission but is not yet approved in the paediatric population
ANCA, anti-neutrophil cytoplasmic antibodies; MRI, magnetic resonance imaging
Challenging cases of ILD
This session discussed some particularly challenging cases and gave some tips on what to look out for:
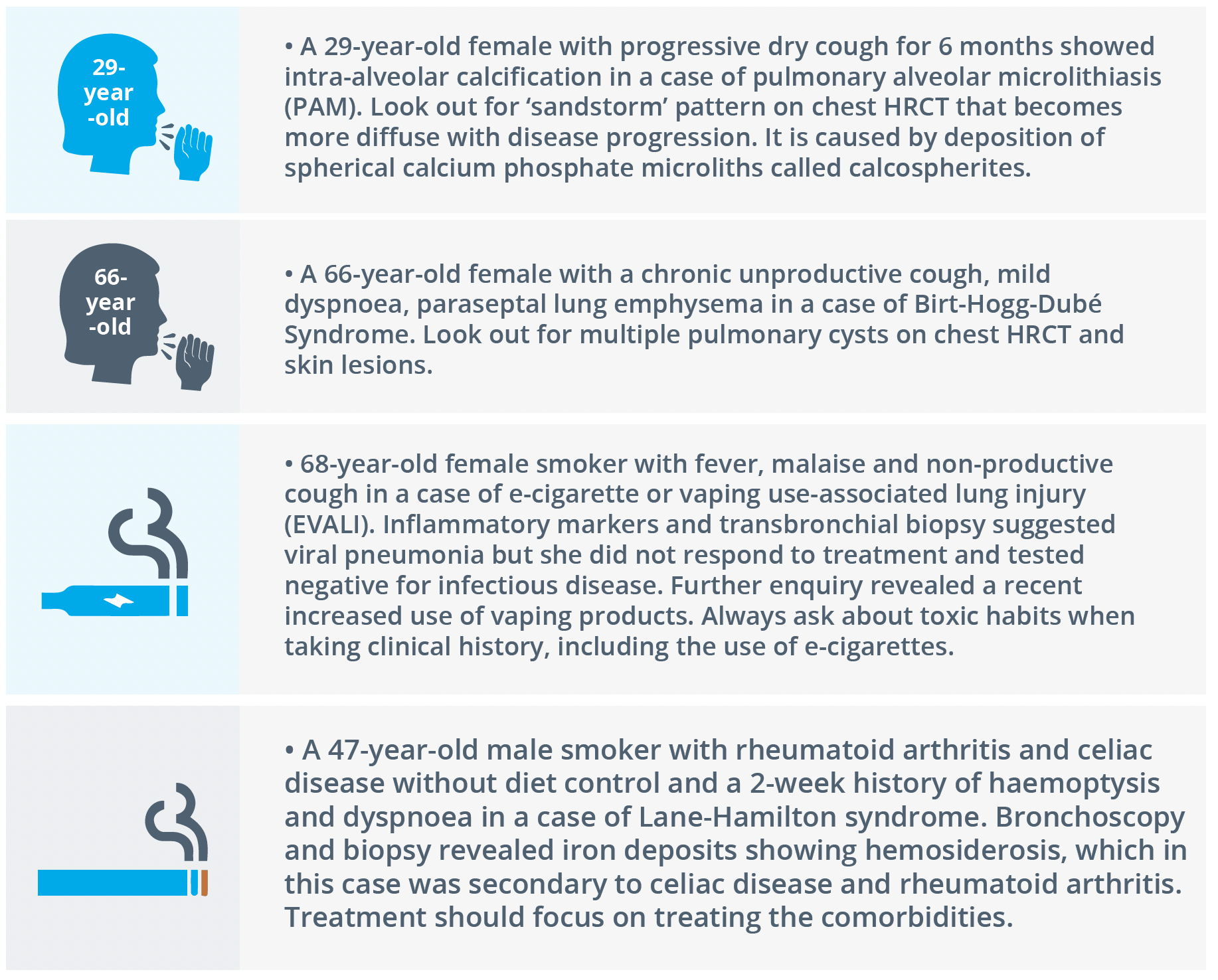
Four distinctive case studies that presented with challenges
ILDs: from disease mechanisms to novel treatment options
In the first presentation of this joint ERS/Lancet session, Elisabetta Renzoni, London, UK, provided a brief review of disease pathology in ILD. There are many different types of lung disease but in essence there are only two ways in which the lung manifests lung injury – inflammation and/or fibrosis. All ILD is therefore characterised by variable degrees of inflammation and fibrosis, not only between diseases, but also among individuals with the same disease. In inflammation-dominant disease, the histology is that of organising pneumonia or NSIP whereas fibrosis-dominant disease would be characterised by UIP. Extensive or progressive fibrotic ILD, regardless of initial pathology, is associated with a high likelihood of progression.

Molecular profiles and immunohistochemistry could refine ILD diagnosis
Histology is key to an accurate diagnosis. Recent developments suggest that molecular profiles and immunohistochemistry could refine the histopathological diagnosis of ILD, providing prognostic markers and potential treatable targets.

Potential biomarkers in ILD
Gisli Jenkins, London, UK, discussed biomarker discovery, development, and implementation in ILD. Biomarkers can be used for different purposes from diagnosis of disease to predicting response to treatment. HRCT remains the ‘gold standard’ diagnostic biomarker for established disease but serum biomarkers may help in early or complex cases. MMP7, either alone or in a panel of different biomarkers, has been shown to correlate with reduced lung function and mortality so may be useful for prognostic assessment. Shortened telomere length, may be useful as an endotypic biomarker. CA-125 is a standardised assay and may have value as a theranostic biomarker.
“MMP7 and CA-125 are almost ready for ‘prime time’ use in the management of IPF”
Gisli Jenkins, London, UK
Diagnostic delay is a common problem in patients with fibrotic ILD, including IPF. Paolo Spagnolo, Padua, Italy, explained that this is due to the insidious onset of the disease and its non-specific, and often minimal, symptoms, as well as low awareness amongst non- ILD experts. The detection of ‘velcro-type’ crackles may offer the most effective means for the earlier diagnosis of IPF. These are fine crackles that are softer, shorter in duration and higher in pitch than coarse crackles. Digitally-recorded lung sounds have been shown to directly correlate with the extent of distinct radiological features of PF. Another approach to improving early diagnosis would be a screening programme for fibrotic ILD but this would currently only be feasible in selected populations at high risk of developing the disease. These would include cases of familiar ILD and patients with connective tissue disease, rheumatoid arthritis, systemic sclerosis or polymyositis, or individuals who have had occupational exposure to hazardous materials.
In the final presentation, Kerri Johannson, Calgary, Canada, reviewed current approaches and future directions of treatment of progressive fibrosing ILD. This should be considered as a clinical phenotype rather than a diagnosis. IPF should be treated with antifibrotics from the outset, whereas fibrotic ILDs that are not IPF should be managed initially with immunomodulatory drugs but will require antifibrotics at later stages of the disease. Non- ILDs include CTD-ILD and fibrotic hypersensitivity pneumonitis (HP).

Disease trajectory with progressive fibrosis
Lung cancer - fibrotic ILD (LC-fILD): a challenging association for pulmonologists
Lung cancer is a comorbidity of ILD. In the first presentation, Manuela Funke-Chambour, Bern, Switzerland, explained that the risk for lung cancer is increased in patients with IPF and that risk has been shown to increase over time. Risk factors include older age at IPF diagnosis, smoking and emphysema.
Typical presentation is squamous cell carcinoma (SQCC) in the lower lobes of the lung. Prognosis is poor in these patients with worse survival in LC-fILD compared to fILD alone.
“There might be an overlap of the mechanisms that underlie the disease pathogenesis”
Manuela Funke-Chambour, Bern, Switzerland
Venerino Poletti, Bari, Italy, discussed the close association of the pathophysiology between lung cancer and fILD. He suggested that the histological and immunophenotypic features of lung cancer developing in patients with IPF show consistent differences from non-IPF carcinomas, with a prevalence of entities showing bronchiolar features.
Treatment strategies in stages I-IV lung cancer typically include combinations of surgery, chemotherapy and radiotherapy. Since there are greater risks of treatment in patients with comorbid LC-fILD, Jacques Cadranel, Paris, France, proposed an adapted table of treatment recommendations. The key differences in treating comorbid LC-fILD are:
- Adjuvant chemotherapy is not recommended in early stages
- Location of the tumour to the fibrotic zone and the presence of emphysema must be considered prior to lung resection due to risk of acute exacerbation (AE)
- Conventional radiotherapy should be avoided due to the risk of radiation pneumonitis
- Carboplatin plus weekly paclitaxel is the standard first-line standard therapy in stage IV
- Addition of an antifibrotic drug may improve outcome and reduce risk of AE

Standard/option recommendations for the treatment and management of NSCLC-fILD
In the final presentation, Athol Wells, London, UK, discussed the use of antifibrotic agents to disrupt malicious LC-fILD pathways. Antifibrotic therapy is warranted in LC-fILD if there is a definite or probable diagnosis of IPF, or if disease is fibrotic and either extensive or progressive even if the diagnosis is uncertain. Data show that antifibrotic therapy can improve ILD outcomes. It has been postulated that they may also improve outcomes by protecting against AE with lung cancer treatments. It has also been hypothesised that they might be able to confer anti-tumour effects.

©Springer Healthcare 2021. This content has been independently selected and developed by Springer Healthcare and licensed by Roche for Medically. The topics covered are based on therapeutic areas specified by Roche. This content is not intended for use by healthcare professionals in the UK, US or Australia. Inclusion or exclusion of any product does not imply its use is either advocated or rejected. Use of trade names is for product identification only and does not imply endorsement. Opinions expressed do not reflect the views of Springer Healthcare. Springer Healthcare assumes no responsibility for any injury or damage to persons or property arising out of, or related to, any use of the material or to any errors or omissions. Please consult the latest prescribing information from the manufacturer for any products mentioned in this material.