
Welcome to HSG 2021

Shari Kinel, CEO of the HSG
Shari Kinel, CEO of the HSG, New York, USA, welcomed all HSG 2021 delegates and thanked the HSG annual planning committee members, executive team and staff. The arrival of HSG’s new contract research organisation was announced, and is comprised of HSG Ltd., a not-for-profit parent company, and HSGCR Inc., a for-profit wholly-owned subsidiary, which runs the operations of HSG’s clinical trials. The total net profit of the clinical trials will be reinvested into HSG, allowing HSG to develop and provide further Huntington’s disease (HD) programs.

Dr Andrew Feigin, HSG Chair
HSG Chair, Dr Andrew Feigin, New York, USA, recounted HSG’s recent clinical trial successes, including PROOF-HD (NCT04556656), a large phase III trial which assessed functional outcomes associated with pridopidine, and the phase III KINECT-HD study (NCT04102579), the first trial of wearable sensors in HD. Dr Feigin also stated that the new HSG Board will be appointed in July 2022.

Elise Kayson, HSG Co-Chair
Elise Kayson, HSG Co-Chair, New York, USA, noted that since the founding of HSG in 1993, HSG has conducted 38 HD clinical trials and studies, including Project AWARE and SIGNAL (NCT02481674). The conduction of such studies led to the approval of the only two FDA-approved drugs available for the treatment of HD, deutetrabenazine and tetrabenazine, demonstrating HSG’s great successes in HD research.
HD is an autosomal dominant neurodegenerative disorder which predominantly presents with chorea, hyperkinesis and executive dysfunction in adult-onset HD. Juvenile-onset HD (<21 years) presents with hypokinetic characteristics with prominent bradykinesia. Pathological features of HD include loss of striatal medium spiny neurons due to mutant Huntingtin (HTT), cortical neuronal loss affecting a variety of neuron types including pyramidal cells and interneurons, and damage to cerebellar cells, particularly the Purkinje cells and neurons in the deep nuclei.

Keynote speaker Jean Paul G Vonsattel, New York, USA, shared insights into the processing and characterisation of post-mortem human brains to investigate the pathogenesis of HD. Dr Vonsattel described his team’s process of post-mortem brain freezing which facilitates the preservation of the neostriatum, with particular focus on standard brain blocks #6 and #7, key areas in HD.

Process of post-mortem brain freezing to facilitate the preservation of the neostriatum (brain blocks SBB6 and SBB7)
Conventional methods include freezing samples with liquid nitrogen and isopentane. However, such methods cause brain blocks to fragment, inhibiting the comparison of the dorsal medial area of the caudate nucleus with the nucleus accumbens. However, the novel method described by Dr Vonsattel uses liquid nitrogen vapour between -180 to -160°C in order to achieve minimal freezing artefacts within brain blocks taken from fresh brain samples.
Dr Vonsattel also discussed atrophic patterns of HD in post-mortem brains relative to those of healthy control brains and the role of medium and large neostriatal neurons in HD and Alzheimer’s disease (AD). In cases of combined HD and AD, enhanced atrophy of the hippocampus and amygdala is often observed. To test whether less striatal atrophy is present in brains with both HD and AD relative to those with HD alone, Dr Vonsattel’s team examined 1201 brains with AD or Lewy body variant AD, 886 with HD and 18 with HD + AD. Brains from individuals with HD alone were matched to the HD + AD group for expanded CAG repeat size, age and gender. Findings demonstrated that the incidence of AD in the HD population resembles that of the general population, and that when HD is present with AD, the two diseases retain their respective phenotypes.
HD genetics
In their 2021 study, Lesley Jones, Cardiff, UK, and her team investigated genetic effects in HD by exome sequencing the age of onset extremes in a cohort of approximately 500 individuals with HD. Their findings identified that repeated CAACAG sequences following the pure CAG tract were associated with an increase in age of onset (+17.9 years, p=6.38 x 10-6). However, the absence of CAA codons following the pure CAG tract was associated with an 18.2-year decrease in age of onset (p=4.84 x 10-6).

Repeated CAACAG sequences following the pure CAG tract has been associated with an increase in age of onset
In addition, they carried out a study which investigated the deoxyribonucleic acid damage and repair effects associated with FAN1 and MSH3. In FAN1, they identified 28 damaging variants and one loss of function (LoF) variant in individuals with early-onset HD, in contrast with 11 damaging variants in those with late onset (p=0.013), indicating segregation across the protein.
In MSH3, little segregation in damaging variants was observed, with LoF variants only in those with late-onset HD. However, this effect was non-significant (p=0.139) due to a small sample size. These findings are consistent with two 2021 studies which demonstrated that the interaction of FAN1 and MLH1 is critical to the promotion of expansions, with FAN1 nuclease activity critical to the specific expansion of the CAG repeat.
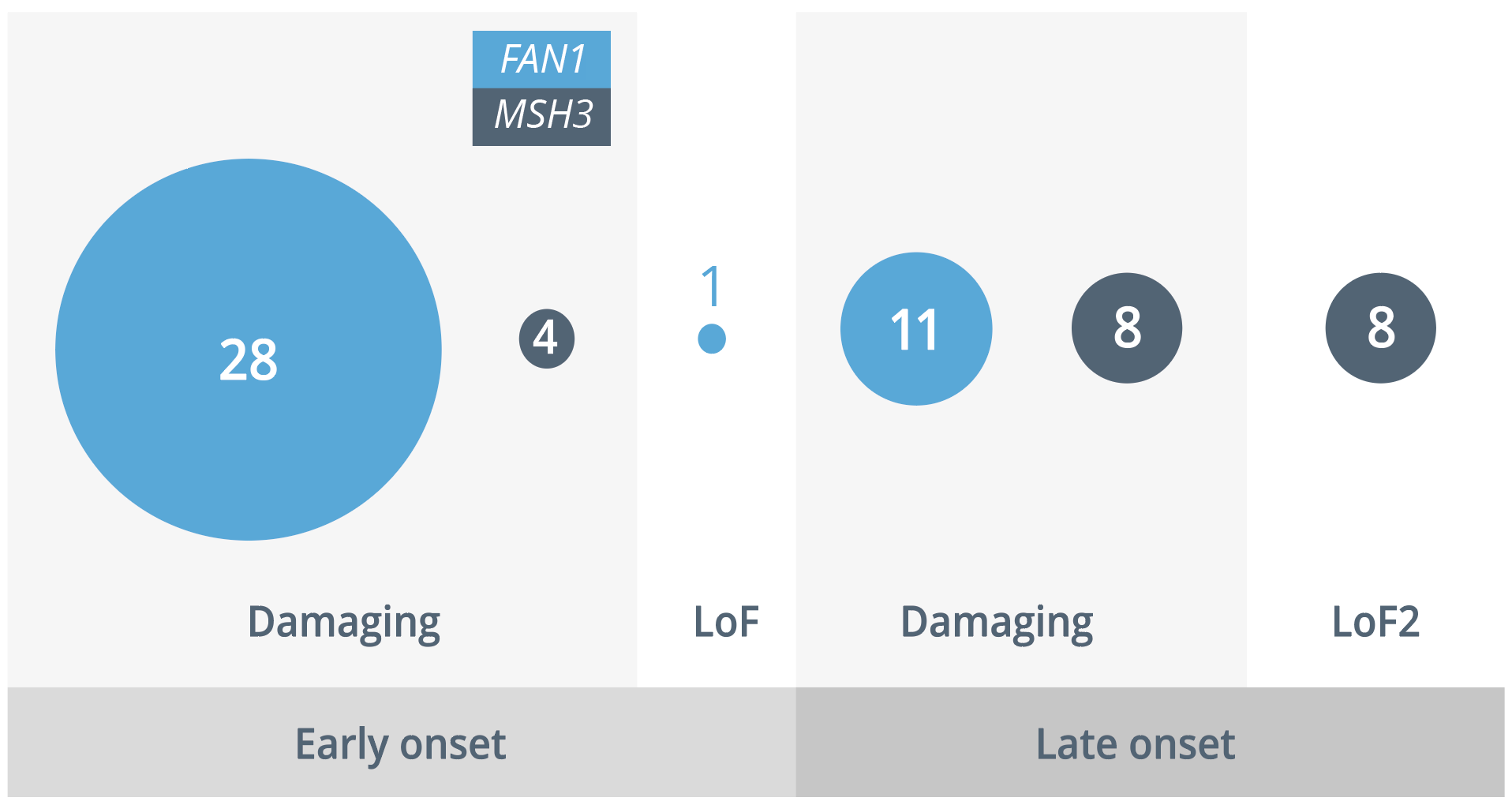
Damaging and loss of function variants in individuals with early- and late-onset HD
Sara Picó, Madrid, Spain, presented findings relating to cytoplasmic polyadenylation element binding (CPEB) protein alterations and aberrant transcriptome-polyadenylation in HD. CPEB proteins are ribonucleic acid (RNA)-binding proteins and recognise transcripts with CPE sequences with a ‘U’-rich motif in their 3’ untranslated region (UTR). Their primary function is to repress or activate polyadenylation-induced translation via shortening or elongation of the poly(A) tail. Bioinformatics approaches demonstrate that approximately 40% of genes are regulated by CPEBs and there is evidence linking CPEBs to cancer, autism and diabetes. However, the potential role of CPEBs in neurodegenerative disorders is yet to have been explored.
Their study sought to explore the status of CPEB4 in HD. Using Western blot analyses, they confirmed an increase in CPEB1 and decrease in CPEB4 levels in post-mortem striatal tissue in patients with HD and subsequently extracted RNA from control and R6/1 mice striatum followed by poly(U) chromatography. Following a genetic analysis, HD, AD and Parkinson’s disease (PD) were identified to be the most significant terms, suggesting that altered polyadenylation may contribute to the pathogenesis of neurodegenerative disorders. Other genes mutated in PD, tauopathies, amyotrophic lateral sclerosis, HD and familial forms of AD also showed polyadenylation, indicating a novel molecular mechanism in the aetiology of neurodegenerative disorders.
Finally, following CPE sequence analyses in the 3’ UTR of genes showing altered polyadenylation, enrichment was observed selectively in those genes showing deadenylation. Western blot analyses confirmed reduced protein levels of the most highly deadenylated genes, including AUTS2, KTN1 and SLC19A3 both in HD and in R6/1 striatal tissue. Interestingly, SLC19A3 mutations are known to cause biotin-thiamine-responsive basal ganglia disease (BTBGD), a neural disorder with prominent striatal affectation which can be improved with thiamine and biotin treatment. In a subsequent preclinical analysis, biotin and thiamine combined therapy prevented the decreased striatal content of TPP in two HD mouse models, and significantly prevented the thiamine deficiency observed in HD mice brains, thus attenuating their phenotypes. Such findings may indicate a novel mechanism in the pathogenesis of HD.
“HD patients might benefit from biotin and thiamine supplementation therapy, as BTBGD patients do.”
Sara Pico, Madrid, Spain
Findings in clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas)-mediated gene therapy in HD were presented by Wenzhen Duan, Maryland, USA. In a study published this year, two guiding RNAs targeting human HTT exon1 regions containing CAG repeats were designed. These were engineered to a viral delivery vector with Cas9, which were delivered to nQ175 mouse brain models to express human HTT therefore mimicking several aspects of HD symptoms. The murine models were used to investigate the treatment effects in altering HD progression. After 9 months, the treated mice performed almost identically to control mice on a balance beam motor task, indicating that CRISPR/Cas9 mediated HTT-lowering may be efficacious in improving motor function in HD mice.
In another 2021 study, the research team engineered mice carrying human single nucleotide polymorphisms (SNP), using CRISPR-Cas9 targeted to SNP areas in order to achieve allele-specific lowering of mutant HTT (mHTT). Findings demonstrated that they were able to achieve allele-specific lowering of mHTT by targeting a protein-coding sequence containing a common, heterozygous SNP, promoting an allele-specific reduction of mHTT in an HD mouse model.
Kate Foreman, North Carolina, USA, discussed why genetic counselling for HD can be hard to access for patients in the USA. The high costs of predictive HD testing in the USA may deter patients as well as genetic counsellors from setting up an initial protocol. Despite HD testing becoming increasingly affordable, the current average total cost to the patient is approximately $11,000 in the USA. Furthermore, due to the complexity of the genetic testing process, which includes many stages such as genetic counselling, neurological examinations and return of results appointment, genetic counsellors may feel they are unable to describe the specific costs to the patient, which may act as a further barrier. In addition, genetic counsellors with limited clinical exposure to HD may lack confidence in implementing protocols in HD genetic counselling. Finally, the limited number of genetic counselling clinics inhibits access for patients, with many having to travel a number of hours to access clinics. Notably, despite the COVID-19 pandemic and the consequent reduction in in-person appointments, patients do not tend to report dissatisfaction with a return of results appointment via telephone in general, resulting in minimal poor outcomes.

The advantages and disadvantages of telehealth for patients with HD during the COVID-19 pandemic
HD pathogenesis
Variations in CAG repeat length can cause significant differences in HD severity and age of onset, both inversely related to CAG repeat length. This trinucleotide repeat occurs 10–35 times within the gene in individuals without HD, however, those with ≥36 repeats are at increased risk of developing the disorder.
CAG repeat ranges in HD

CAG repeat ranges from 9-40+ and the effect increasing penetrance has on HD risk
Tim Koscik, Iowa, USA, proposed an HD pathogenesis framework which synthesises the five primary pathogenic mechanisms of HD. A multitude of factors have been demonstrated to contribute to the pathogenesis of HD, described in this framework, including CAG repeat expansions, abnormal cortical development and neuropathology.

HD pathogenesis network from inherited CAG repeat expansion to brain network effects
“Taken altogether, this framework suggests that in order to improve our understanding of HD pathology, we need to explore the whole brain to capture widespread brain networks; we need to look in sufficient detail at features that capture pathological development.”
Tim Koscik, Iowa, USA
Dr Koscik also discussed the limitations of in vivo brain imaging, which include insufficient resolution, human factors limiting scan time and excessive physiological movement, such as micro-movements caused by cerebral blood pressure. Cellular and molecular imaging methods, despite providing sufficient detail, are limited by their laborious, invasive and destructive nature. Therefore, post-mortem imaging may circumvent current limitations in exploring the HD pathology. With these methods, ultra-high resolution images of entire brains can be achieved, with the absence of motion during scan times. In addition, brain tissue remains intact due to virtual slicing, and reconstruction into an entire brain can be encoded.

Eight-step process of assessing post-mortem samples in patients with juvenile-onset HD
HD treatment
Results from the Physical Activity and Exercise Outcomes in HD (PACE-HD) study (NCT03344601) were presented by Monica Busse, Cardiff, UK. PACE-HD was a multi-centre cohort trial carried out at sites across Europe and the USA and sought to evaluate long-term physiotherapist-led physical activity outcomes in patients with early-to-mid stage HD. A total of 120 participants were recruited to the trial: 60 in the reference cohort, who received no contact or intervention, and 60 enrolled in a randomised controlled trial. All patients were followed up for 12 months, with those enrolled in the randomised clinical trial undergoing an additional 6-month interim assessment. Participants were provided a face-to-face coaching intervention and an HD-specific physical activity workbook, as well as optional fitness equipment or gym funding. The study was based on self-determination theory, promoting autonomy, competence and relatedness.
Autonomy |
Competence |
Relatedness |
• Freedom of choice |
• Perceived self-belief |
• Sense of shared experience |

Overall, no serious adverse events were reported by either group and fall incidence was similar in both RCT groups.
“Consultative models of care which involve support for a long-term physical therapist-led physical activity intervention should be routine in HD clinical care.”
Monica Busse, Cardiff, UK
Jee Bang, Maryland, USA, discussed new and upcoming research in HD therapeutics. Current therapeutic approaches focus on two main outcomes: neuroprotection (e.g., pridopidine) and HTT protein-lowering using RNA interference (e.g., AMT-130) or anti-sense oligonucleotides (e.g., WVE-003).

Upcoming clinical trials include the evaluation of SAGE-718, an oral solution which modulates activity of the N-methyl-D-aspartate (NMDA) receptor. A small preliminary study demonstrated improvements in cognitive testing outcomes in patients with early HD; testing of which is planned to continue in a future phase II study. Upcoming gene therapies include branaplam, an oral drug which lowers HTT protein levels in mice and children with spinal muscular atrophy, and TTX-3360 which targets MSH3 and thus reduces somatic expansion.
The role of complementary and integrative medicine (CAIM) in HD was discussed by Danny Bega, Illinois, USA. Current conventional HD treatments at times incite an inadequate response, causing adverse effects or not effectively treating symptoms such as irritability and motor difficulties. Further, the paucity of disease-modifying treatments for HD has led patients and families to seek out alternative options to current conventional care. A 2010 study found that 44.1% of 6587 US adults with neurological conditions reported using CAIM in the previous 12 months, often in parallel with conventional treatment. Examples of CAIM include natural products, such as herbal medicines and vitamins, mind-body practices including acupuncture and massage, and alternative systems, such as Chinese medicine and homeopathy. Overall, CAIM therapies appear to have a good safety profile and low-cost. However, the lack of scientific evidence supporting CAIM treatments must be noted, and caution exercised when combining these therapies with other drugs.

Ten most commonly used CAIM approaches in the USA (N=88,962) from guided imagery (1.7%) to natural products (17.7%)
“There is something to be gained by not just being a passive recipient of care, but actually being an active participant in your day-to-day life and in your health.”
Danny Bega, Illinois, USA
Management of HD
Katherine McDonell, Tennessee, USA, provided an overview of the behavioural symptoms associated with HD. In general, HD symptoms fall into three main categories:

Psychiatric/behavioural symptoms are often the most burdensome for patients and caregivers. However, such symptoms are often easily treatable, thus significantly improving patients’ and caregivers’ quality of life.

A major concern involves suicide risk in HD, with suicides rates at 5–6 times those in the general population (world population risk = 1.5%; risk in patients with HD = 4.8–6.6%). In addition, suicide is the second most common cause of death in HD, the risk of which appears to be particularly marked at the time of initial symptom onset and when loss of independence begins.
Management of suicide risk
Other behavioural symptoms of HD include irritability, perseveration, apathy, obsessive/compulsive symptoms, delusions or hallucinations and impulsivity. Symptoms which require immediate attention are aggressive behaviour, suicidal thoughts, physical threats or thoughts of harming others and severe delusions or hallucinations. The concept of human flourishing in medicine was discussed by Tyler VanderWeele, Massachusetts, USA. The principle domains of flourishing include happiness and life satisfaction, physical and mental health, meaning and purpose, character and virtue and close social relationships. Each of these domains are nearly universally desired, and exist as an end in themselves.


Primary questions to assess flourishing on a scale of 1-10
The measurement of human flourishing can prove useful in evaluating the wellbeing of an individual or population. To this end, Professor VanderWeele and his team collected data on the ten domains above in January 2020, prior to the World Health Organisation declaration of the COVID-19 pandemic, and subsequently in June 2020. As expected, scores decreased overall; however, each of the domains were affected differently. For instance, financial and material stability suffered the highest mean score loss (-0.9/10), while smaller losses were seen in meaning and purpose (-0.4/10) and close social relationships (-0.2/10), with character and virtue scores remaining unchanged. Close social relationships scores decreased less than expected, perhaps due to individuals finding ways to connect virtually with friends and family.
Flourishing has also been observed to affect physical health, with recent studies finding that purpose in life and life satisfaction reduce mortality risk (relative risk [RR] = 0.83, CI = 0.75–0.91; RR = 0.88, CI = 0.83–0.94, respectively), while loneliness and social isolation increase mortality risk (RR = 1.29, CI = 1.06–1.56).
Closing remarks

Dr Andrew Feigin, Elise Kayson and Shari Kinel, New York, USA, thanked all presenters, sponsors and delegates for contributing to and attending the 2021 annual HSG meeting, with hopes that HSG will return to its in-person format in 2022.
©Springer Healthcare 2021. This content has been independently selected and developed by Springer Healthcare and licensed by Roche for Medically. The topics covered are based on therapeutic areas specified by Roche. This content is not intended for use by healthcare professionals in the UK, US or Australia. Inclusion or exclusion of any product does not imply its use is either advocated or rejected. Use of trade names is for product identification only and does not imply endorsement. Opinions expressed do not reflect the views of Springer Healthcare. Springer Healthcare assumes no responsibility for any injury or damage to persons or property arising out of, or related to, any use of the material or to any errors or omissions. Please consult the latest prescribing information from the manufacturer for any products mentioned in this material.