Pemphigus Vulgaris (PV): Treatment & Management
Clinical criteria for PV diagnosis
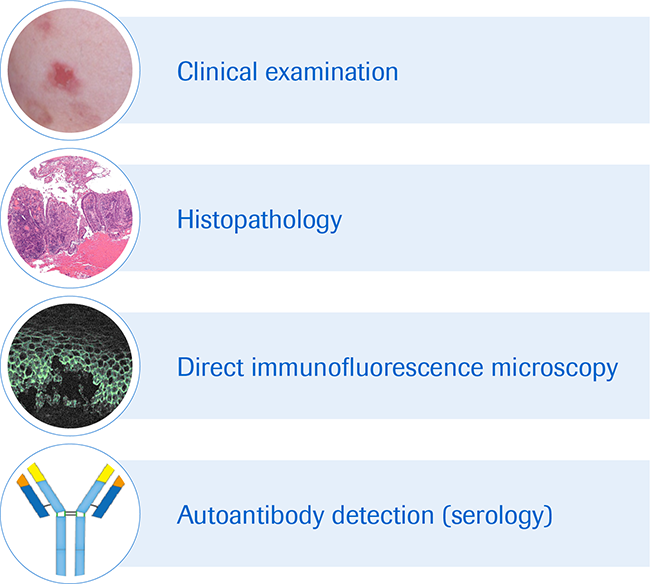
Diagnosis of PV involves a clinical examination (medical history and physical examination) and a series of laboratory investigations to confirm PV.1
In the physical examination, the characteristics and extent of skin and mucous membrane lesions, and the degree of mucosal damage and functional impairment is assessed. The extent and distribution of lesions in PV can be quantified using one of the two validated scoring systems, either the Pemphigus Disease and Area Index (PDAI) or the Autoimmune Bullous Skin Intensity and Severity Score (ABSIS).1
Laboratory assessments are then undertaken using skin biopsies and blood samples to confirm that it is PV and not another blistering or erosive disease. These assessments include detecting active acantholysis (histopathology), autoantibodies at the surface of epidermal keratinocytes (direct immunofluorescence microscopy) and circulating autoantibodies (serology).1
- Histopathology: A small biopsy (3-5 mm punch excision) of affected skin (blister and/or lesion) is taken and examined for evidence of acantholysis in the basal layer by histopathology. Basal keratinocytes remain attached to the basement membrane but not each other, creating a characteristic ‘tombstone effect’.
- Direct immunofluorescence (DIF) against cell surface antigens: A skin biopsy (1 cm of fresh lesion) is examined for the presence of IgG autoantibodies to DSG3 and DSG1 and possible complement C3 protein deposits on the surface of epidermal keratinocytes.
- Serology to detect serum autoantibodies: B-cell produced anti-DSG1 and/or anti-DSG3 antibodies are measured in serum of patients with PV by enzyme-linked immunosorbent assay (ELISA). ELISA testing is positive in more than 95% of PV cases. Serum concentrations for anti-DSG diagnosed by ELISA may help in therapeutic decision making as anti-DSG levels correlate with the activity and extent of disease. An alternative test is indirect immunofluorescence (IIF) microscopy, in which anti-epithelial cell surface IgG deposits can be detected on the epithelium of monkey or guinea pig esophagus or human skin when exposed to serum from a PV patient. Antibody titers broadly correlates with the disease activity and/or the extent of disease.
Criteria for clinical diagnosis of PV
PV may be first seen by general practitioners, dentists, oral surgeons or gynecologists, depending on where the symptoms first appear. Rapid referral to a specialist dermatologist is important to ensure early diagnosis. PV is treatable and prompt intervention may prevent the development of more severe symptoms.
PV treatment & management
The goal of PV treatment is to dampen the autoimmune response to prevent development of new blisters and to promote healing of existing blisters and lesions.
Objectives of PV treatment
PV is an autoimmune blistering disease driven by the production of auto-antibodies against DSG1 and DSG3. Most therapies aim to improve symptoms through the reduction of serum autoantibodies, either by targeted therapy or through generalized immune suppression.2
The primary objective of PV treatment is to control and heal the bullous skin and/or mucosal lesions, whilst minimizing the side effects of treatment. PV is usually managed by a specialist dermatologist with experience in treating bullous diseases.4
Treatment aims:4
To promote healing of blisters and erosions
To improve functional status
To prevent/strictly limit development of new blisters and erosions
To improve patient’s quality of life
To minimize the common side-effects usually associated with long-term immunosuppressive or corticosteroid treatment
PV treatment and management guidelines
Current guidelines:
EADV/EDF guidelines for the management of patients with pemphigus1
The Autoimmune blistering diseases Task Force of the European Academy of Dermatology and Venereology (EADV) has published guidelines for the management of patients with pemphigus. The final version of the guideline was consented by the European Dermatology Forum (EDF) and several patient organizations.
For the therapeutic management of PV, the guideline provides recommendations for initial and maintenance treatment for mild and moderate to severe PV.
International recommendations for the diagnosis and management of pemphigus4
To broaden the generalizability of the EADV/EDF guidelines across the globe, the International Bullous Diseases Consensus Group developed international consensus recommendations for the diagnosis and treatment of pemphigus.
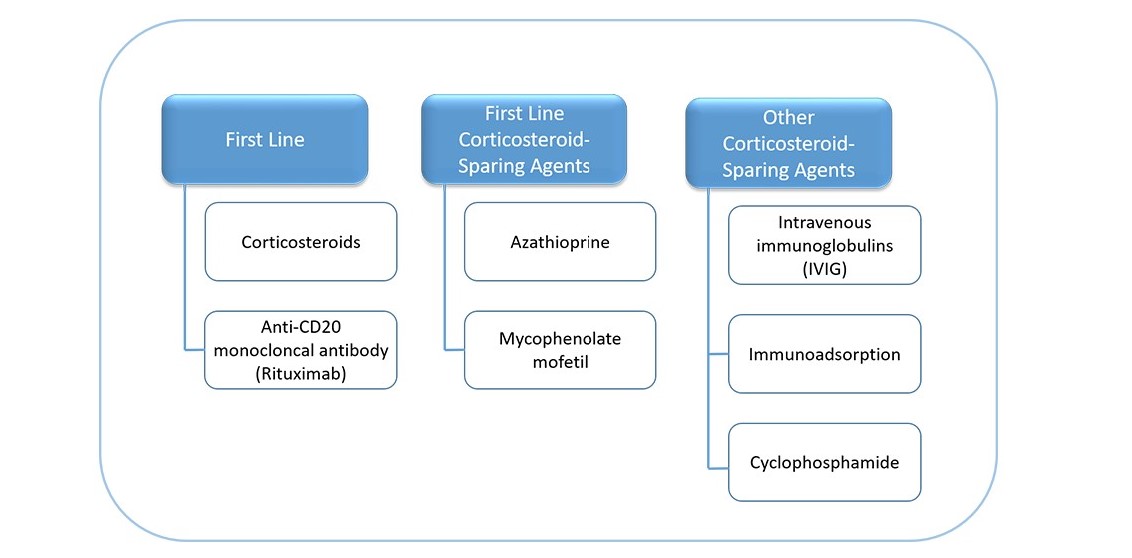
- Systemic corticosteroids can be combined with an immunosuppressive adjuvant at the onset of therapy, especially in cases of increased risk of corticosteroid therapy, complications due to expected prolonged use (>4 months), or dose dependency above minimal therapy (>10mg/day). First-line corticosteroid-sparing agents listed are azathioprine and mycophenolate mofetil or mycophenolic acid, other corticosteroid-sparing agents include intravenous immunoglobulins, immunoadsorption or cyclophosphamide (figure below).
- Anti-CD20 monoclonal antibody (rituximab) is recommended for first-line treatment in new onset moderate to severe pemphigus and/or for patients who do not achieve clinical remission with corticosteroids and/or immunosuppressive adjuvants.
PV treatment options2
Once patients have started treatment, it is important to monitor the efficacy and safety of the treatment and to plan the gradual reduction of immunosuppressive treatment and the duration of maintenance therapy or its discontinuation. In an initial phase, the treatment aim is that patients achieve disease control. Disease control is achieved once no new blisters appear for 2 weeks and most existing lesions have healed. This marks the end of the consolidation phase of therapy and usually marks a change in the therapeutic management. This is when most clinicians start to taper steroids1,4
After the consolidation phase, patients need to be monitored for the level of disease control, presence of adverse effects due to treatment, and disease activity (determination of serum autoantibodies). The frequency of consultations depends on the patient’s clinical condition including comorbidities, the disease severity, therapeutics used, and level of disease activity.1,4
The consensus group also provides recommendations on supportive treatment, such as proper dental care, intralesional injections of corticosteroids, topical treatment with potent corticosteroids, antiseptic baths, analgesics, local anesthetics and nutritional management, and prophylaxis against side effects in prolonged corticosteroid therapy.
References
- Joly P, et al. J Eur Acad Dermatol Venereol. 2020, 34, 1900-1913
- Kasperkiewicz M, et al. Nat Rev Dis Primers. 2017; 3:17026.
- Hebert V, et al. J Invest Dermatol. 2018; doi: 10.1016/j.jid.2018.1004.1042.
- Murrell DF, et al. J Am Acad Dermatol. 2020; 82(3): 575–585