
Pathophysiology
PNH Causes
PNH is caused by a somatic mutation in the phosphatidylinositol glycan class A (PIGA) gene. The PIGA gene encodes a glycosyl transferase, a crucial enzyme in the biosynthetic pathway responsible for generating glycosyl phosphatidylinositol (GPI). The mutations in the PIGA gene lead to a loss of enzyme function, resulting in diminished expression of all GPI-anchored proteins. Two essential complement inhibitory proteins that are affected by the PIGA mutation are CD55 and CD59. Thus, PNH differs from other haemolytic anaemias in two ways: it involves the haematopoietic stem cell (HSC) rather than just erythrocytes, and its faulty process is acquired, not inherited.1
Complement physiology
The complement system is part of innate immunity, a system that is activated in response to infection, inflammation or trauma. These events trigger one of three pathways that can initiate the complement cascade to enhance the initial signal and create a strong response:
- Classical pathway (detects antibody-antigen immune complexes)
- Lectin pathway (recognises carbohydrates on the surface of pathogens)
- Alternative pathway (constitutively active at low levels)2,3
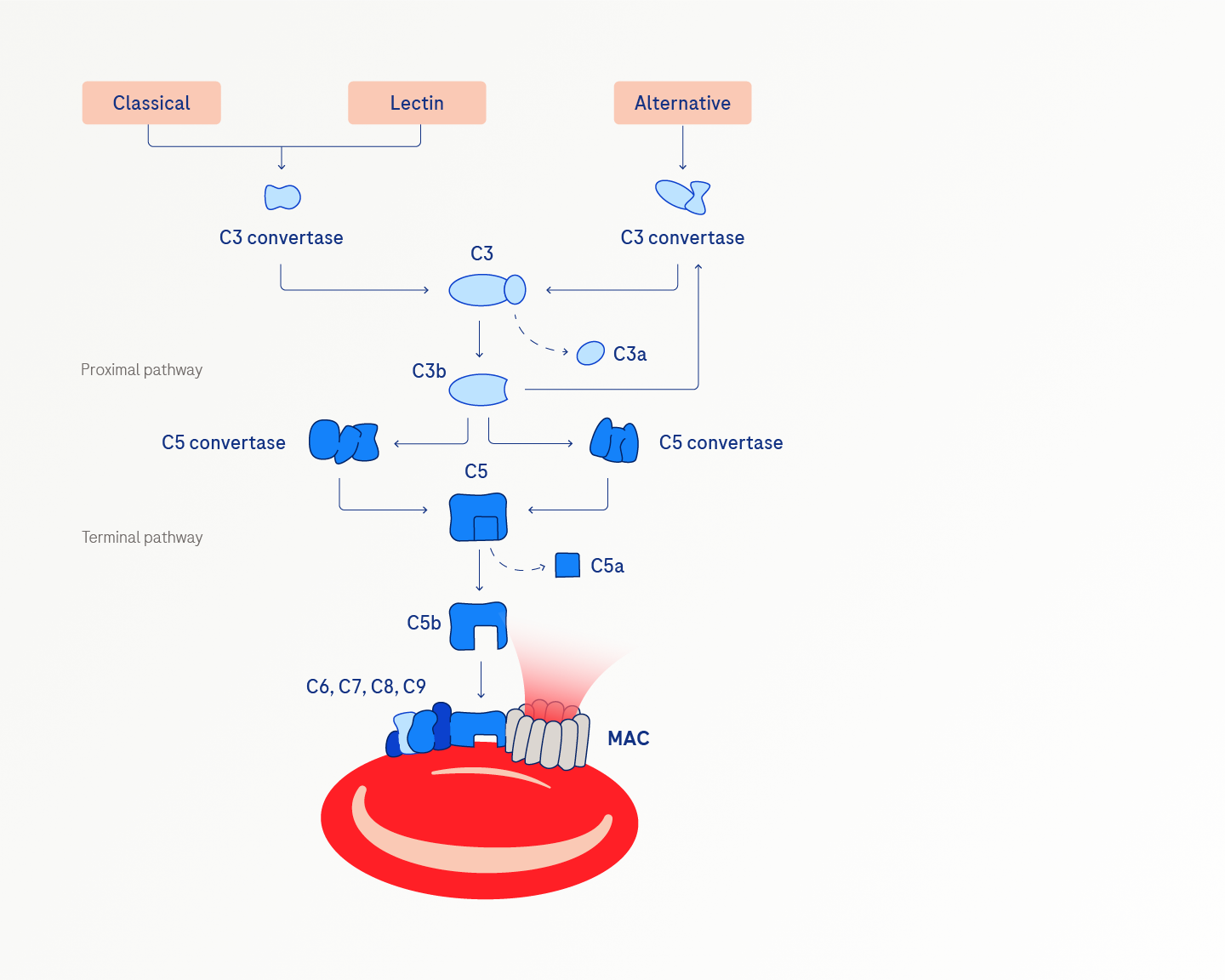
The terminal pathway is common to the three complement activation pathways. The terminal effector of the complement system is the membrane attack complex (MAC) that creates pores in the pathogenic cell membranes, causing them to burst or lyse.2,3
Complement is normally tightly regulated by several mechanisms, one of which involves regulatory proteins on the cell surface that prevent C5 activation and MAC formation. Disruptions in GPI synthesis due to the PIGA mutation interfere with the normal complement-inhibiting function of CD55 and CD59, which underlies the complement-mediated intravascular haemolysis of PNH.3
Consequences of impaired GPI-anchoring
PNH develops from the clonal expansion of PIGA-mutated haemopoietic stem cells in the bone marrow. HSCs differentiate into multiple blood cell types, producing a population of cells with the PNH phenotype called PNH clones.4
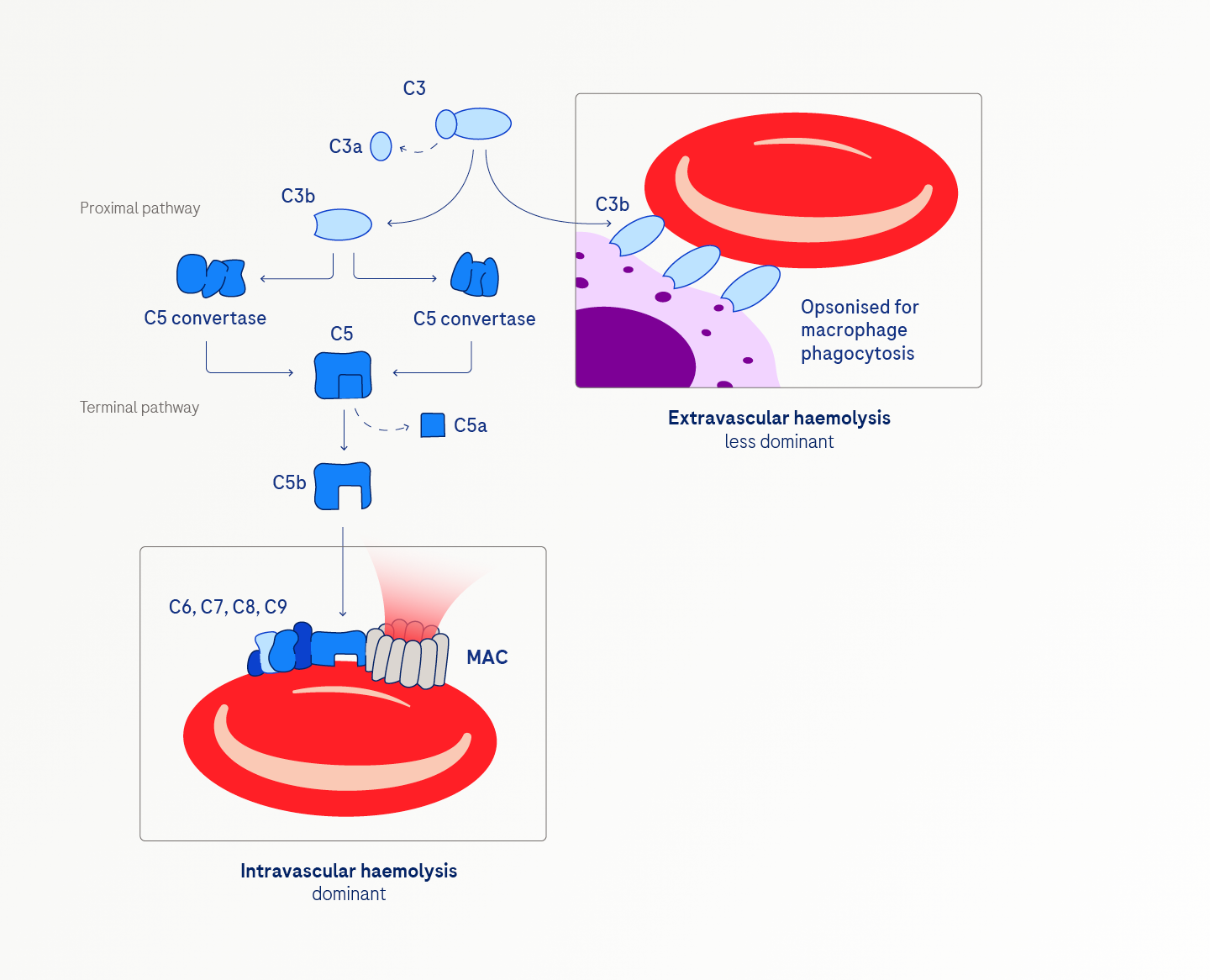
The consequences of impaired GPI anchoring are multifaceted. Intravascular haemolysis, the predominant feature, is marked by the complement-mediated lysis of red blood cells due to uncontrolled MAC-formation. Red blood cells are the most vulnerable, but platelets and white blood cells are also affected.4,5,6
Extravascular haemolysis, though a lesser contributor, can occur and may be unmasked in patients receiving terminal complement inhibitors. This process involves persistent, uncontrolled activation of the proximal complement, leading to C3 accumulation and the opsonisation of PNH red blood cells by C3. Phagocytes in the liver and spleen then clear and destroy these opsonised cells.5,6,7
PNH clones can be found in patients with other bone marrow failure syndromes, like aplastic anemia and many patients with PNH have some degree of underlying bone marrow failure. Unlike intravascular haemolysis and thrombosis, bone marrow failure in PNH is not a downstream event of the PIGA mutations; rather, it independently results from cellular autoimmunity to HSCs.4
Learn more about how to diagnose PNH
and explore other PNH resources
References:
- Parker CJ. Update on the diagnosis and management of paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program. 2016; 2016 (1): 208–216. doi:10.1182/asheducation-2016.1.208
- Janeway CA Jr, Travers P, Walport M et al. Immunobiology: The Immune System in Health and Disease. 5th edition. Garland Science; New York, 2017
- Duval A, Frémeaux-Bacchi V. Complement biology for hematologists. Am J Hematol. 2023; 98 (Suppl 4): S5–S19. doi:10.1002/ajh.26855
- Hill A, DeZern AE, Kinoshita T et al. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. 2017; 3: 17028. doi:10.1038/nrdp.2017.28
- Bessler M, Hiken J. The pathophysiology of disease in patients with paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program. 2008; 104–110. doi:10.1182/asheducation-2008.1.104
- Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014; 124 (18): 2804–2811. doi:10.1182/blood-2014-02-522128
- Risitano AM, Peffault de Latour R. How we('ll) treat paroxysmal nocturnal haemoglobinuria: diving into the future. Br J Haematol. 2022; 196 (2): 288–303. doi:10.1111/bjh.17753