
Huntington's Disease Hub: Medical Resources
What is Huntington’s disease?
Huntington's disease (HD) is a rare, monogenic neurodegenerative disease characterised by a triad of cognitive, behavioural and motor symptoms leading to functional decline and progressive loss of independence.1-4 It typically strikes in the prime of life, between 30 and 50 years of age,3 and impacts families across generations, with each child of a parent with HD having a 50/50 chance of developing the disease.1 There is currently no proven approach to slowing or stopping the relentless progression of this ultimately fatal disease.2
The more we learn about the mutant huntingtin (mHTT) protein responsible for causing HD, and how we may be able to lower its levels, the closer we get to unlocking the mysteries of this condition.

HD is the most common monogenic neurological disorder in the developed world. HD has an average prevalence rate of ~10 in 100,000, which has increased worldwide between 9–20% each decade.5-9
Watch to learn more about HD
Signs & symptoms of HD
HD is characterised by a triad of cognitive, behavioural and motor symptoms. It leads to functional decline and loss of independence,3,4 and results in death on average 15 years after the onset of motor symptoms.10
Cognitive disturbances in HD can occur years before diagnosis and onset of motor symptoms, and deteriorate steadily as the disease progresses. Behavioural manifestations in HD are particularly diverse and can also occur many years before a clinical diagnosis of HD is made.11-14 Motor symptoms in HD are initially subtle and progress in a non-linear trajectory over the course of the disease.4,15

Since individuals experience these symptoms in their own unique way, HD can often be difficult to diagnose. This means it can also be challenging for patient care and a multidisciplinary approach may help improve disease management.
*This is not a comprehensive list of HD symptoms. Symptoms and signs differ for each individual with HD.3
Stages of HD
HD is a continuum which can be described in three stages. Clinical diagnosis of HD is typically defined by the onset of unequivocal motor symptoms and occurs when people are in the prime of life, typically between the ages of 30 and 50 years.3,4,17,18 HD phasing terminology continues to evolve; however, the disease generally progresses through the presymptomatic, prodromal and manifest stages.1,3,18
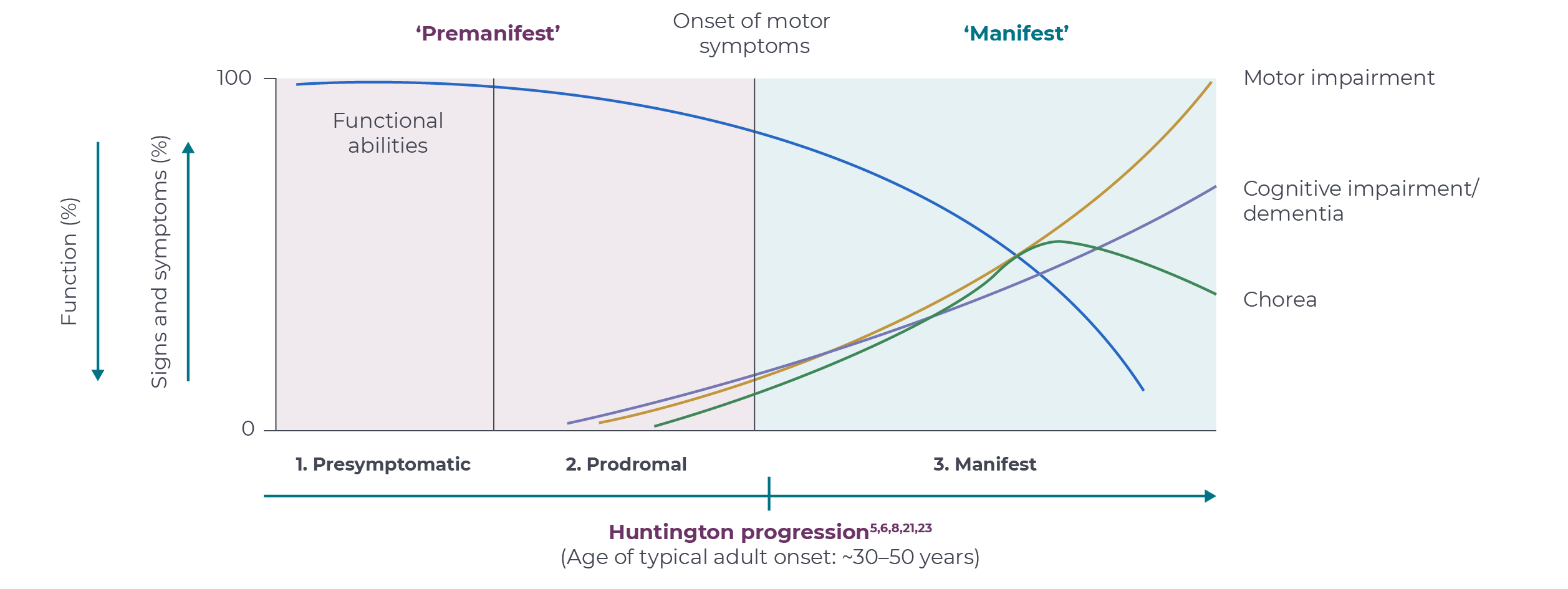
1. Presymptomatic: Individuals who carry the HD-causing gene mutation but have not yet developed any symptoms.1,2,4
2. Prodromal: Individuals experience subtle changes in cognition, mood and behaviour that appear years before diagnosis or onset of unequivocal motor signs.2,4,17,19 Brain changes, including striatal atrophy, are apparent.2
3. Manifest: Individuals with HD have unequivocal motor symptoms and are clinically diagnosed with HD.2,4
Learn more about Huntington’s disease pathology & mechanisms of disease
References
1. Ghosh R & Tabrizi SJ. Huntington disease. In Handbook of Clinical Neurology, vol. 147 2018; pp. 255–278. Edited by Geschwind DH, Paulson HL & Klein C. Elsevier BV.
2. Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers. 2015; 1:15005.
3. Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010; 5:40. doi:10.1186/1750-1172-5-40.
4. Ross C, Aylward E, Wild E, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014; 10:204–216.
5. Rawlins M, Wexler N, Wexler A, et al. The Prevalence of Huntington’s Disease. Neuroepidemiology. 2016; 46:144–153.
6. Evans S, Douglas I, Rawlins M, et al. Prevalence of adult Huntington's disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry. 2013; 84:1156–1160.
7. Squitieri F, Griguoli A, Capelli G, et al. Epidemiology of Huntington disease: first post-HTT gene analysis of prevalence in Italy. Clin Genet. 2016; 89:367–370.
8. Fisher E & Hayden M. Multisource Ascertainment of Huntington Disease in Canada: Prevalence and Population at Risk. Mov Disord. 2014; 29:105–114.
9. Rawlins M. Huntington's disease out of the closet? Lancet. 2010; 376:1372–1373.
10. Keum J, Shin A, Gillis T, et al. The HTT CAG-Expansion Mutation Determines Age at Death but Not Disease Duration in Huntington Disease. Am J Hum Genet. 2016; 98:287–298.
11. Paulsen JS, Long JD, Ross CA, et al. Prediction of manifest Huntington's disease with clinical and imaging measures: a prospective observational study. Lancet Neurol. 2014; 13:1193–1201.
12. Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013; 12:637–649.
13. Paulsen J. Cognitive Impairment in Huntington Disease: Diagnosis and Treatment. Curr Neurol Neurosci Rep. 2011; 11:474–483.
14. Rosenblatt A. Neuropsychiatry of Huntington's disease. Dialogues Clin Neurosci. 2007; 9:191–197.
15. Paulsen J, Long J, Johnson H, et al. Clinical and Biomarker Changes in Premanifest Huntington Disease Show Trial Feasibility: A Decade of the PREDICT-HD Study. Front Aging Neurosci. 2014; 6:78.
16. Anderson KE, van Duijn E, Craufurd D, et al. Clinical management of neuropsychiatric symptoms of Huntington disease: expert-based consensus guidelines on agitation, anxiety, apathy, psychosis and sleep disorders. J Huntingtons Dis. 2018; 7(3):355–366.
17. Reilmann R, Leavitt BR, Ross CA. Diagnostic criteria for Huntington's disease based on natural history. Mov Disord. 2014; 29:1335–1341.
18. Ross CA, Reilmann R, Cardoso F, et al. Movement Disorder Society Task Force Viewpoint: Huntington's Disease Diagnostic Categories. Mov Disord Clin Pract. 2019; 23;6:541–546.
19. Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008; 79:874–880.