
Huntington's Disease Treatments Hub
New frontiers in HD treatment
Novel disease-modifying therapies are under investigation in HD
HD is caused by a cytosine–adenine–guanine (CAG) trinucleotide repeat expansion in the huntingtin gene (HTT) resulting in the production of a toxic mutant HTT (mHTT) protein.1-4 mHTT causes progressive neuronal degeneration, ultimately leading to neuronal cell death.2,4,5 Levels of mHTT in cerebrospinal fluid correlate with disease stage, symptom severity and markers of neuronal damage in people with HD.5
Novel disease-modifying therapies are being investigated in both pre-clinical and clinical studies, exploring different approaches to slow or stop progression of this debilitating disease.6-9
Disease-modifying therapies under investigation include antisense oligonucleotides (ASOs), gene therapies and small molecule splice modifiers:
1. Antisense oligonucleotides (ASOs)
Antisense oligonucleotides (ASOs), administered intrathecally, have the potential to change the therapeutic landscape for many neurological conditions including HD.7
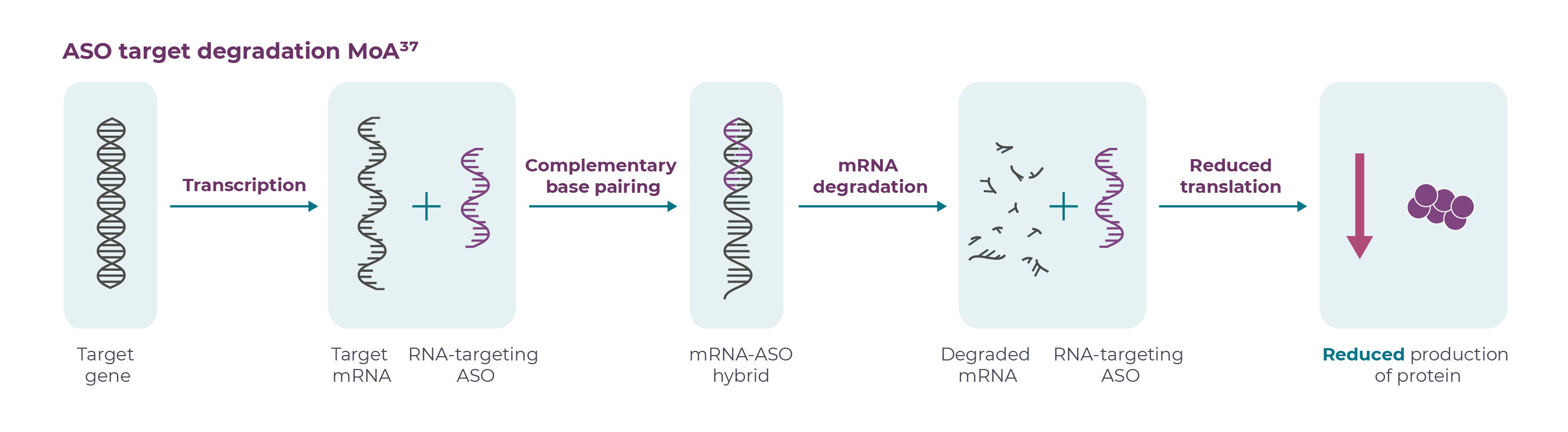
ASOs are short, synthetic strands of nucleic acids, typically between 16–20 nucleotides in length, that bind to target pre-mRNA or mRNA and reduce, modify or restore protein expression.7,8 There are several distinct mechanisms of action of ASOs depending on their chemistry, binding sequence and target.7. In current HD research, once bound to the RNA, ASOs form an RNA-DNA hybrid, and the target mRNA is degraded, preventing translation into protein.7,9
Allele-specific (targeting only the mutant allele) and non-allele specific (targeting both wild-type and mutant alleles) approaches are under investigation for lowering HTT.8,10
2. Gene therapy
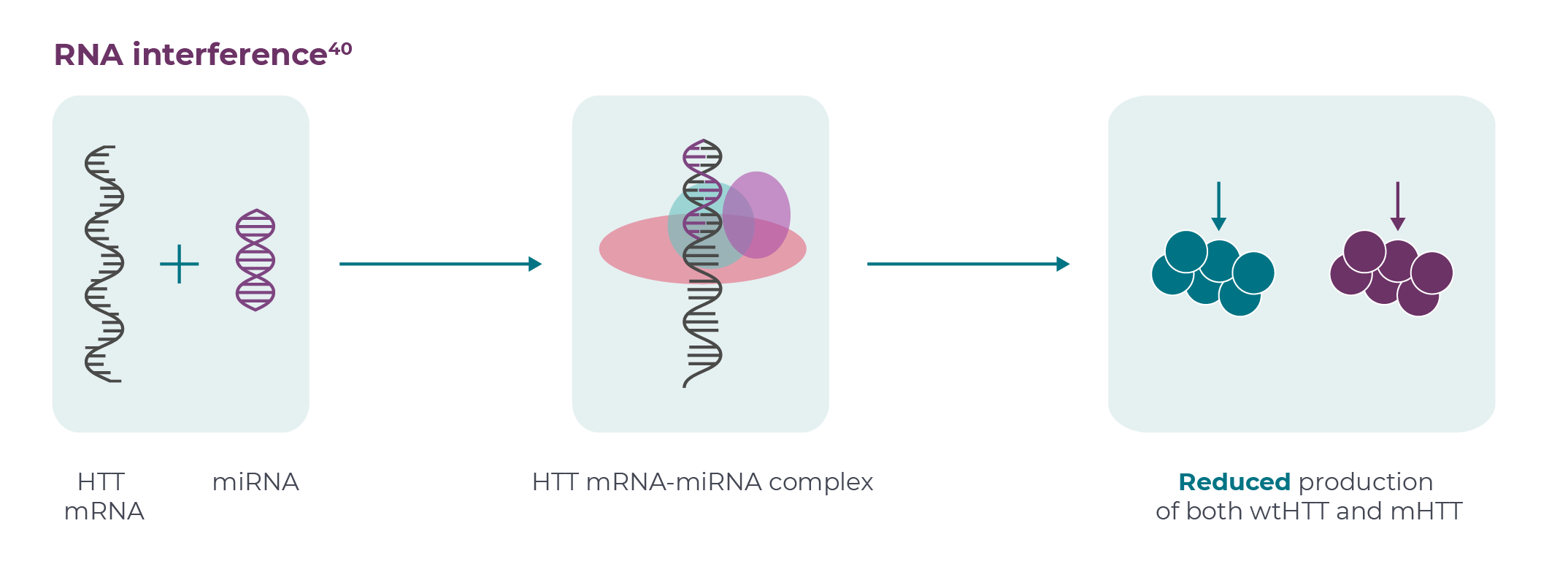
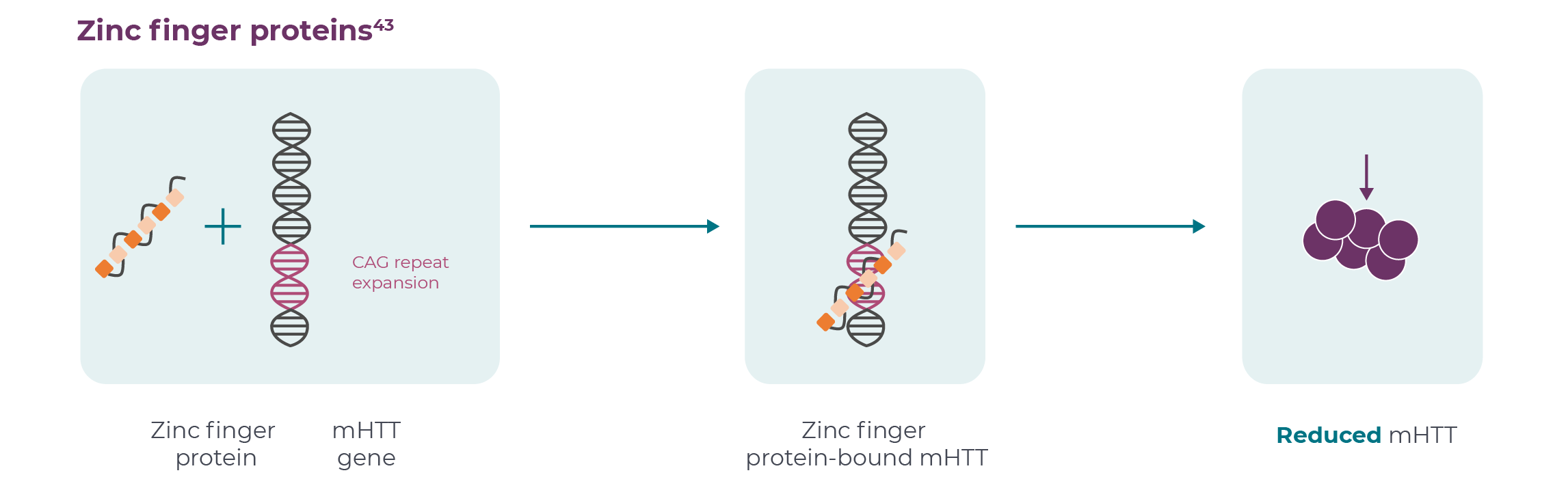
According to the FDA, “Human gene therapy aims to modify or manipulate the expression of a gene or to alter the biological properties of living cells for therapeutic use”.11 There are several approaches; those currently under investigation for HD include one-off intrastriatal administration of genes encoding microRNA (miRNA) or zinc finger proteins (ZFPs).10,12,13 miRNAs are small non-coding RNAs that play important roles in regulating gene expression.14 ZFPs are proteins that can bind DNA and play a role in a range of cellular processes, including development, differentiation and neurodegeneration.13,15
Genetic material-encoding miRNA or ZFPs are delivered using viral vectors,13 which lack viral DNA and are designed to cross the cell membrane to deliver the novel genetic material.16
The miRNA approach suppresses translation through RNA interference (RNAi): binding of miRNA to HTT mRNA transcripts induces their degradation, thereby preventing translation into protein.10
The ZFP approach suppresses transcription of the mHTT gene. ZFPs for HD have been developed to bind selectively to the CAG repeat expansion, and therefore may preferentially suppress transcription of the mHTT allele and reduce production of the mHTT protein.13
3. Small molecule splice modifiers
Pre-mRNAs are new, immature strands of messenger RNA (mRNA) following transcription that contain both introns (non-coding sections) and exons (coding sections).17 RNA splicing is a form of RNA processing in which the newly made pre-mRNA transcript is transformed into a mature mRNA. During splicing, introns are removed and exons are joined together.18 Small molecule splice modifiers can be used to inhibit protein production by altering the splicing of pre-mRNA.10,19,20 These orally bioavailable small molecules can penetrate the blood-brain-barrier and are under investigation in HD to lower levels of mHTT.10,21
To see the latest news in HD research, go to HDBuzz
You can explore ongoing clinical trials in HD and find studies that are actively recruiting participants at HD Trial Finder. Find out more about the ongoing trials in HD
Learn more about new frontiers in HD treatment
References
1. Huntington’s Disease Collaborative Research Group. A novel gene containing trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993; 72:971–983.
2. Ghosh R & Tabrizi SJ. Huntington disease. In Handbook of Clinical Neurology, vol. 147 2018; pp. 255–278. Edited by Geschwind DH, Paulson HL & Klein C. Elsevier BV.
3. Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers. 2015; 1:15005.
4. Saudou F & Humbert S. The Biology of Huntingtin. Neuron. 2016; 89:910–926.
5. Wild EJ, Boggio R, Langbehn D, et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J Clin Invest. 2015; 125(5):1979–1986.
6. National Institute of Neurological Disorders and Stroke, National Institutes of Health. Huntington’s Disease: Hope Through Research. National Institutes of Health website: https://catalog.ninds.nih.gov/pubstatic//17-NS-19/17-NS-19.pdf (Accessed May 2020).
7. Rinaldi C & Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018; 14:9–21.
8. Lane RM, Smith A, Baumann T, et al. Translating Antisense Technology into a Treatment for Huntington's Disease. Methods Mol Biol. 2018; 1780:497–523.
9. Liang XH, Sun H, Nichols JG, et al. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol Ther. 2017; 25:2075–2092.
10. Wild EJ & Tabrizi SJ. Therapies targeting DNA and RNA in Huntington's disease. Lancet Neurol. 2017; 16:837–847.
11. Food and Drug Administration (FDA). Cellular & Gene Therapy Products. FDA website: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products (Accessed May 2020).
12. Wang D & Gao G. State-of-the-art human gene therapy: part II. Gene therapy strategies and clinical applications. Discov Med. 2014; 18:151–161.
13. Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin Lowering Strategies for Disease Modification in Huntington's Disease. Neuron. 2019; 101(5):801–819.
14. O'Brien J, Hayder H, Zayed Y, et al. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne). 2018;9:402.
15. Cassandri M, Smirnov A, Novelli F, et al. Zinc-finger proteins in health and disease. Cell Death Discovery. 2017;3:17071.
16. Naso MF, Tomkowicz B, Perry III WL, et al. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs. 2017; 31(4):317–334.
17. Scitable by Nature Education: Intron/introns. Nature website: https://www.nature.com/scitable/definition/intron-introns-67/ (Accessed May 2020).
18. Pandya-Jones A. Pre-mRNA Splicing During Transcription in the Mammalian System. Wiley Interdiscip Rev RNA. 2011; 2(5):700–717.
19. Sivaramakrishnan M, McCarthy KD, Campagne S, et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun. 2017; 8:1476.
20. Poirier A, Weetall M, Heinig K, et al. Risdiplam distributes and increases SMN protein in both thePoirier A, Weetall M, Heinig K, et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect. 2018; 6:e00447.
21. PTC Therapeutics and CHDI Foundation Announce a Collaboration on a Small-Molecule Therapeutic for Huntington's Disease. PTC Therapeutics website: http://ir.ptcbio.com/news-releases/news-release-details/ptc-therapeutics-and-chdi-foundation-announce-collaboration (Accessed May 2020).