
Huntington's Disease Pathology & Mechanisms of Disease
Pathology & mechanisms of disease
mHTT: The protein with big impact
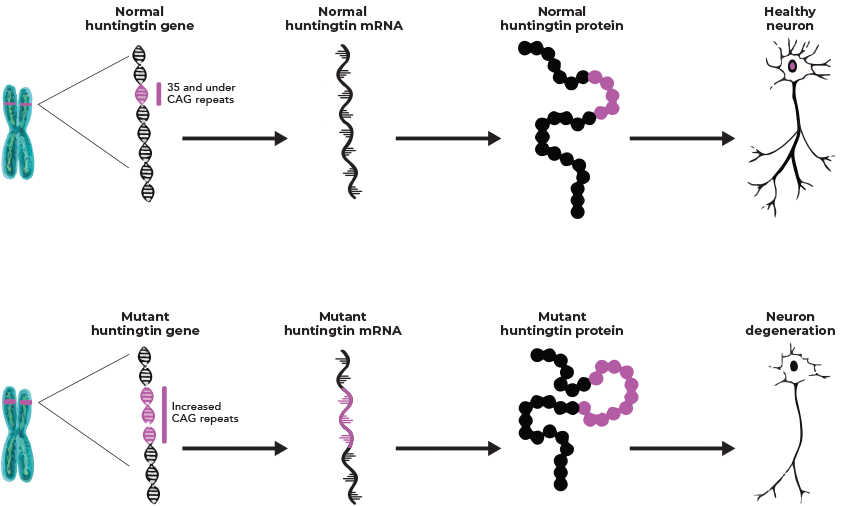
Huntington’s disease (HD) is caused by a cytosine–adenine–guanine (CAG) trinucleotide repeat expansion in the huntingtin gene (HTT); a gain-of-function mutation leading to the production of toxic mutant huntingtin (mHTT) protein.1 The degree of symptom severity, disease stage, and markers of neuronal damage have been shown to correlate with levels of the mHTT protein in the cerebrospinal fluid in individuals with HD. This toxic mHTT causes progressive neuronal degeneration, ultimately leading to neuronal cell death.1-4
The role of the mHTT protein in HD progression
The production of this toxic mutant huntingtin protein leads to progressive neuronal degeneration, ultimately leading to neuronal cell death.5 Levels of mHTT protein in cerebrospinal fluid have been shown to correlate with disease stage, symptom severity and markers of neuronal damage in people with HD.5
The exact mechanism by which mHTT protein causes neuronal death is still being elucidated; however, research suggests that it may interfere with a number of cellular processes such as DNA transcription and axonal transport.6
The significance of expanded CAG repeats
Researchers identified that the number of CAG trinucleotide repeat expansions in the huntingtin gene (HTT) has been shown to correlate with the age of disease onset.2,3 It is known that a CAG repeat length of ≥40 results in definite HD,1,7-10 CAG repeat length inversely correlates with age of onset,3,7 and other genetic and environmental factors may also affect disease progression.3,11
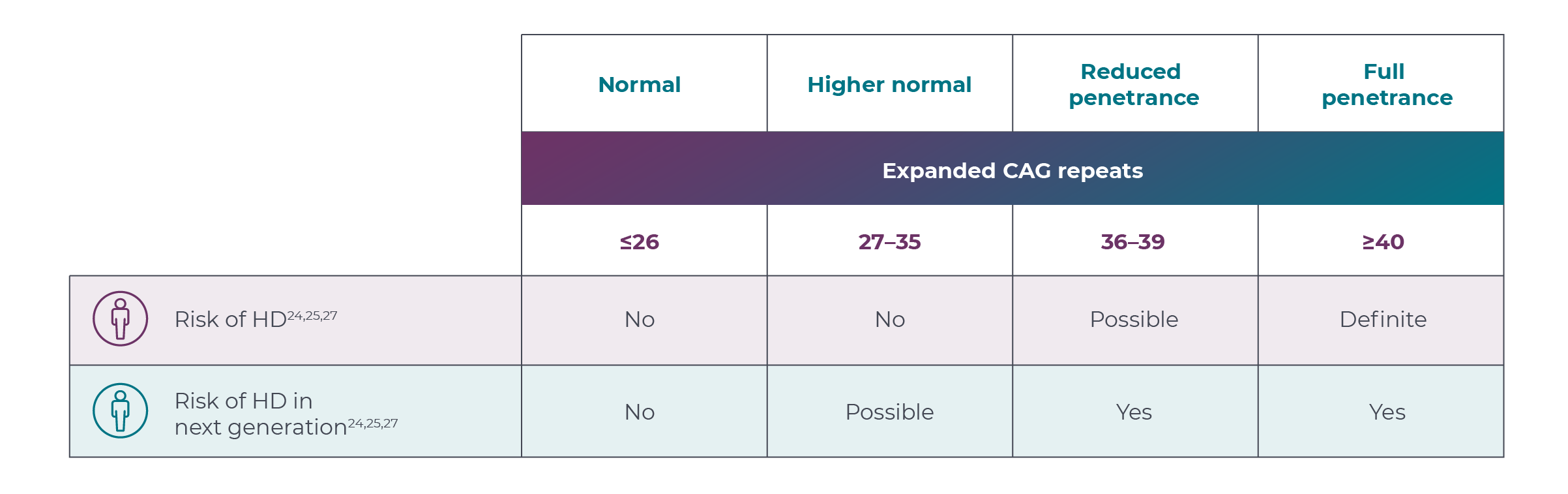
A blood test can be performed to determine the CAG repeat length.2,12 More information can be found on the NIH website.
How is the understanding of the mHTT protein helping further Huntington’s disease research?
Hear from researchers Nancy Wexler and Lauren Byrne on why the identification of the fundamental cause of HD was so significant, and how that finding informs today’s research into potential disease-modifying therapies that may slow or stop disease progression.
References
- Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers. 2015; 1:15005.
- Huntington’s Disease Collaborative Research Group. A novel gene containing trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993; 72:971–983.
- Ghosh R & Tabrizi SJ. Huntington disease. In Handbook of Clinical Neurology, vol. 147 2018; pp. 255–278. Edited by Geschwind DH, Paulson HL & Klein C. Elsevier BV.
- Saudou F & Humbert S. The Biology of Huntingtin. Neuron. 2016; 89:910–926.
- Wild EJ, Boggio R, Langbehn D, et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J Clin Invest. 2015; 125(5):1979–1986.
- Frank S. Treatment of Huntington’s disease. Neurotherapeutics. 2014; 11(1):153–160.
- Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010; 5:40. doi:10.1186/1750-1172-5-40.
- Potter NT, Spector EB, Prior TW. Technical Standards and Guidelines for Huntington Disease Testing. Genet Med. 2004; 6:61–65.
- The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group. Laboratory Guidelines for Huntington Disease Genetic Testing. Am J Hum Genet. 1998; 62:1243–1247.
- Nance M, Paulsen JS, Rosenblatt A, et al. Physician’s Guide to the Management of Huntington’s Disease. 3rd ed 2011. New York, NY: Huntington's Disease Society of America.
- Ross C, Aylward E, Wild E, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014; 10:204–216.
- National Institute of Neurological Disorders and Stroke, National Institutes of Health. Huntington’s Disease: Hope Through Research. National Institutes of Health website: https://catalog.ninds.nih.gov/pubstatic//17-NS-19/17-NS-19.pdf (Accessed May 2020).