In-Depth Report
The 2021 World Muscle Society (WMS) Virtual Congress took place between September 20th and 24th and provided an update on the latest developments in neuromuscular diseases from around the world. WMS comprises a dynamic community that aims to promote, disseminate and share all aspects of neuromuscular physiology and diseases, from basic science to patient care. This year’s virtual congress was centred around a comprehensive scientific programme, delivered through the online platform, which included a mix of state-of-the art oral presentations, debate sessions, e-poster displays and industry-sponsored symposia.
Welcome to WMS 2021

The 2021 congress began with an introduction and welcome by WMS President Volker Straub, Newcastle, UK, who highlighted the great spectrum of translational research topics covered at this year’s meeting – ranging from preclinical developments, new genes and techniques, to clinical research and therapeutic trial highlights. It is thanks to these important advances in translational research that we have already moved from clinical trials to improved patient care for a number of neuromuscular disorders, noted Dr Straub.
“The society’s collaboration continues to be extremely important as safe and effective treatments for patients with neuromuscular diseases can only be developed in close partnership between colleagues from academia, healthcare professionals, industry and – of course – the patient community.”
- Volker Straub, Newcastle, UK
Artificial intelligence applications in the neuromuscular field

The opening keynote lecture entitled ‘Why artificial intelligence (AI) will be the foundation of 21st century medicine’ was delivered by Brendan Frey, Toronto, Canada, and focused on applications of smart technology such as AI and machine learning to the neuromuscular field.
Most breakthroughs in AI over the last decade have been due to deep learning, a type of machine learning that leverages multiple layers of interconnected artificial neurons known as neural networks. Potential applications of AI in biomedicine include the analysis of genetic data and target identification, quantitative structure-activity relationship modelling in drug discovery and biomedical imaging. AI has the ability to address issues of complexity, accuracy and/or scale and can generate new drug targets and molecules, but requires huge amounts of digital data to achieve this. Another key condition for the success of AI in drug discovery is a modular biological framework and in this respect RNA affords an ‘amazing toolkit’, explained Dr Frey. By using the Deep Genomics platform to screen candidate exons and explore the predictive effect of therapies, it proved possible to ‘discover nusinersen for spinal muscular atrophy (SMA) in one afternoon on a computer.’ In contrast to traditional drug discovery approaches, digital AI workbenches enable drug discovery outcomes to be predicted and “derisked” upfront. Data-driven prediction in AI also generates positive feedback loops that drive exponential growth - ‘The more you do, the better the system gets,’ declared Dr Frey. To date, Deep Genomics has unlocked 40 machine learning predictors with potential applications ranging from novel target discovery and therapy design, prediction of safety and toxicity and identification of new patient populations. Deep learning has the potential to unlock disease targets across the spectrum of genetic types and has already progressed from simple Mendelian recessive indications to therapy areas characterised by many complex effects. However, important challenges still lie ahead for AI in medicine, stressed Dr Frey, notably earning the trust of key stakeholders. To achieve this, it is essential to provide causal rationale and explanations for AI outcomes that are amenable to probing and can be discussed and compared.
“RNA therapies are digital information where the sequence of letters determines what the molecule will do … For the first time ever, we are seeing medicines as information,”
- Brendan Frey, Toronto, Canada
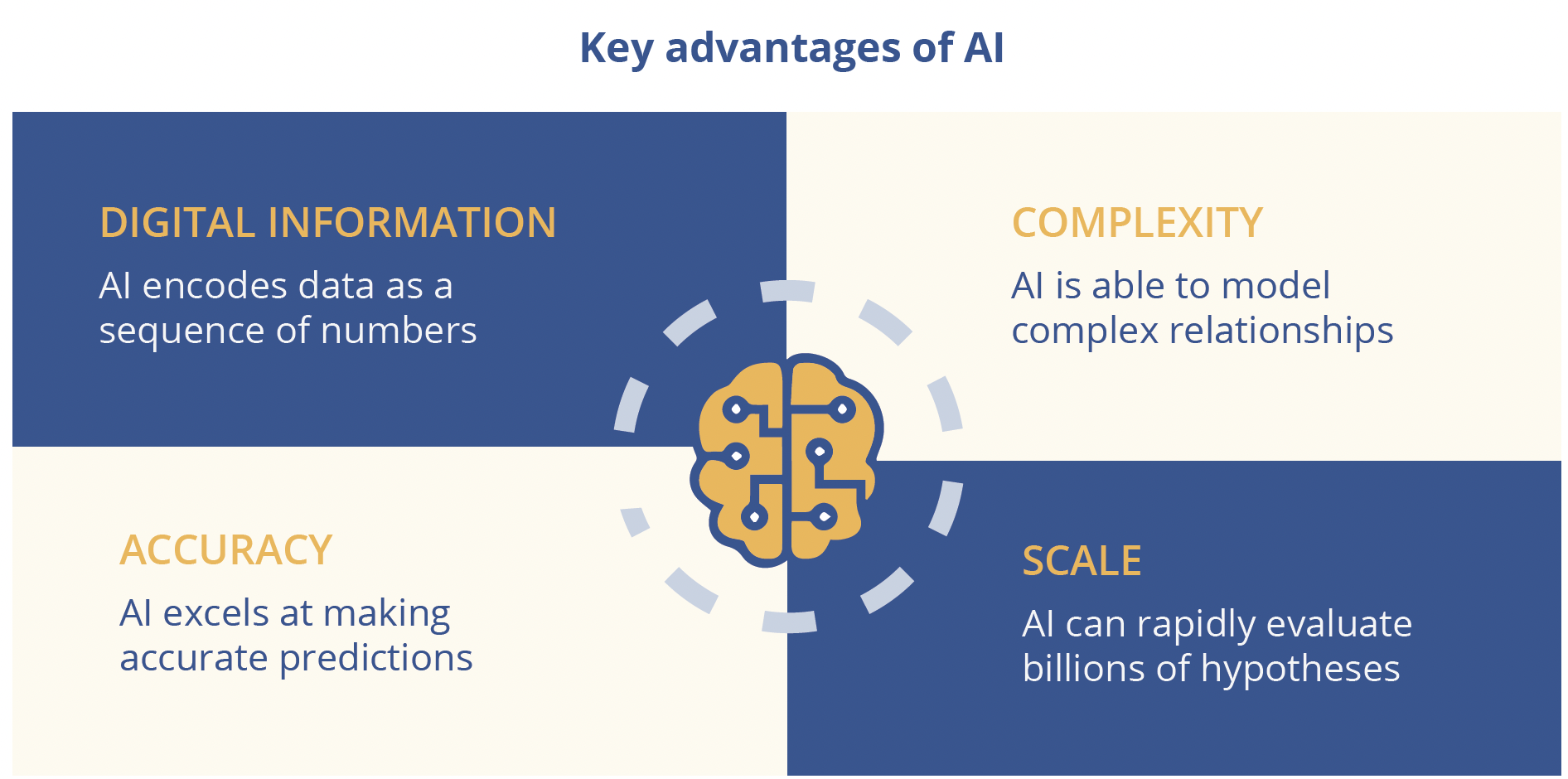
Key advantages of AI include: complexity, digital information, accuracy and scale
Key advances in neuromuscular disease clinical trials
Translational medicine is essential to the field of neuromuscular disorders, bridging the gap between basic science and clinical trials in order to bring new therapeutic options to patients around the world. The Clinical Trial Highlights session showcased key findings from international trials of both gene therapy and pharmacotherapy for the treatment of neuromuscular diseases.
SGT-001 is an experimental gene therapy for Duchenne muscular dystrophy (DMD) which uses an adeno-associated virus 9 (AAV9) vector to deliver a unique, rationally designed microdystrophin that retains the key neuronal nitric oxide synthase (nNOS) binding domain. Varnshi Rao, Chicago, USA, provided an update on long-term outcomes from the IGNITE phase I/II ascending-dose study of SGT-001 (NCT03368742), focusing on data for the three patients in the higher dose 2E14 vg/kg cohort. Muscle biopsy analysis conducted ≥12 months post-administration of SGT-001 showed durable expression and function of microdystrophin protein and limited dystrophic pathology progression. Sustained or increased microdystrophin protein levels and percent positive muscle fibres were observed, together with sarcolemmal restoration of key dystrophin associated proteins β-sarcoglycan and nNOS.

SGT-001 gene therapy produced important improvements compared with natural history over 1.5 years
Meaningful improvements were also seen in patient-reported outcome measures that gauge motor function and fatigue in DMD. Treated patients showed sustained increases from baseline in Paediatric Outcomes Data Collection Instrument (PODCI) scores for sports and physical functioning, as well as transfer and basic mobility. Based on safety data for the entire study cohort (N=8), nausea, vomiting and fever were the most common drug-related clinical adverse reactions associated with SGT-001 gene therapy.

Difference in 6MWT, NSAA and FVC with SGT-001 gene therapy versus natural history over 1.5 years

“The totality of data supports continued dosing in IGNITE DMD at the 2E14 vg/kg dose”
- Varnshi Rao, Chicago, USA
In clinical studies of SMA, the time from symptom onset to therapy initiation has been established as a key predictive factor influencing treatment efficacy. Preliminary data from 5 of 12 infants with presymptomatic SMA currently enrolled in the RAINBOWFISH study of risdiplam (NCT03779334) and treated for
≥12 months were presented by Laurent Servais, Oxford, UK. RAINBOWFISH is a multicentre, open-label, single arm study of the oral survival of motor neuron 2 (SMN2) pre-mRNA splicing modifier, risdiplam. It is enrolling infants up to 6 weeks of age (at time of first dose) with a genetic diagnosis of 5q autosomal recessive SMA but with no clinical signs or symptoms at screening. The primary endpoint of the trial is the proportion of infants able to sit without support for ≥5 seconds at Month 12 (Bayley Scales of Infant Development [BSID]-III Gross Motor Scale, Item 22).
At the data cut-off for this interim analysis, 80% of risdiplam-treated infants (4 out of 5) scored the maximum Hammersmith Infant Neurological Exam Part 2 (HINE-2) total score of 26, which included one infant with two SMN2 copies. One infant with two SMN2 copies had a HINE-2 total score of 23. Four out of five infants treated for ≥12 months with risdiplam achieved standing and walking independently within the World Health Organisation (WHO) windows for healthy children (6.9–16.9 and 8.2–17.6 months, respectively) and all five infants reached the maximum score of 64 on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND; as of data cut-off). Furthermore, all five infants who received at least 1 year of risdiplam therapy maintained the ability to swallow and were able to feed exclusively by mouth. No treatment-related serious adverse events (SAEs) were reported in presymptomatic infants treated with risdiplam for up to 18.1 months and AEs were more reflective of the age of infants than the underlying SMA (e.g. nasal congestion, teething etc). Importantly, no ophthalmic, haematologic or dermatologic clinical safety findings were observed in any infant treated in RAINBOWFISH. The RAINBOWFISH study is continuing to recruit globally, confirmed Dr Servais, and will help to determine the optimal dose of risdiplam for use in infants less than 2 months of age. Target enrollment is 25 patients.
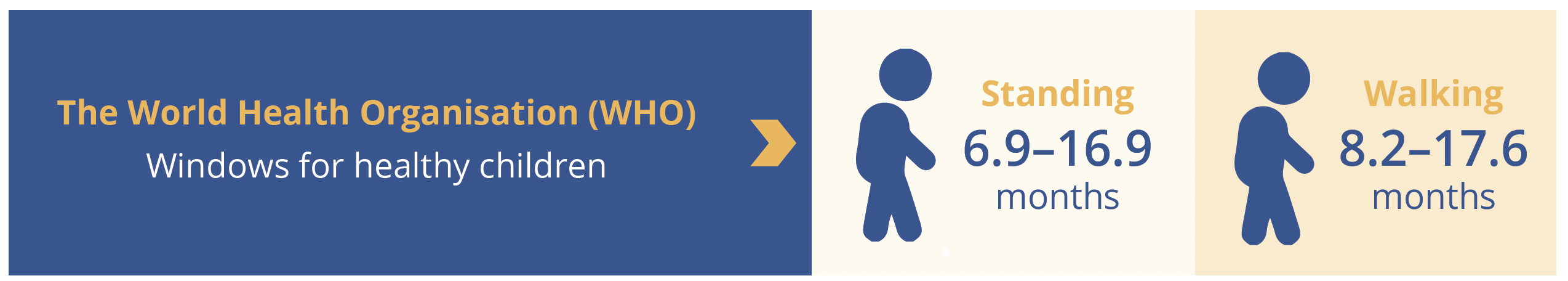
WHO windows for standing and walking independently for healthy children
Exploring natural history in neuromuscular disorders

This clinical research session focused on the importance of a well-characterised natural history, including genotype-phenotype correlations and validated outcome measures, as a fundamental building block in the design of clinical trials for new therapies for neuromuscular disorders.
Charlotte Lilien, Oxford, UK, reviewed findings from a prospective natural history study of upper limb disease evolution in DMD. Despite the large number of studies conducted to date, further exploration of the natural history of DMD is still warranted, Dr Lilien explained. This is due to a lack of prospective, longitudinal studies in upper limb disease in ambulatory patients and the need to better explore severe phenotypes. It is also important to understand the impact of new, recently approved treatment options such as golodirsen and viltolarsen on disease evolution. This 7-year prospective longitudinal study, a collaborative effort between the UK and France, enrolled a total of 40 patients eligible to exon 53 skipping followed-up for a minimum of 3 years. In 2015, 4 years into the study, a protocol amendment was introduced to include only non-ambulatory patients treated with glucocorticoids. Strength and functional assessments were performed every 6 months and quantitative magnetic resonance imaging (MRI) carried out once per year of forearm flexor and extensor muscles to measure fat fraction and lean muscle cross-sectional area. The resulting patient population was well balanced between ambulatory (n=18) and non-ambulatory groups (n=22) and had a mean age of 11.7 years.
Results showed a continuous evolution of disease over time, discriminated between ambulatory and non-ambulatory patients, but with individual variability. At 24 and 36 months follow-up for non-ambulatory patients, standardised response means (SRMs) were significant (>0.8) for almost all outcomes with the exception of Walton score and MoviPlate score. This study was therefore able to identify sensitive outcome measures that can detect changes over a 3-year period in non-ambulatory DMD patients with high SRM, Dr Lilien observed. Correlation analysis revealed that the relationship between strength and function was not linear, but exponential. The relationship between strength and fat fraction also showed an exponential decrease, meaning that patients’ strength dramatically decreased with the onset of fat fraction infiltration. In an attempt to decipher clinically meaningful predictors for patients, the study combined two measures - grip strength and fat fraction. The resulting model proved reproducible at baseline and Year 3; Year 1 and Year 4 and, Year 2 and Year 5 for both ambulatory and non-ambulatory patients.
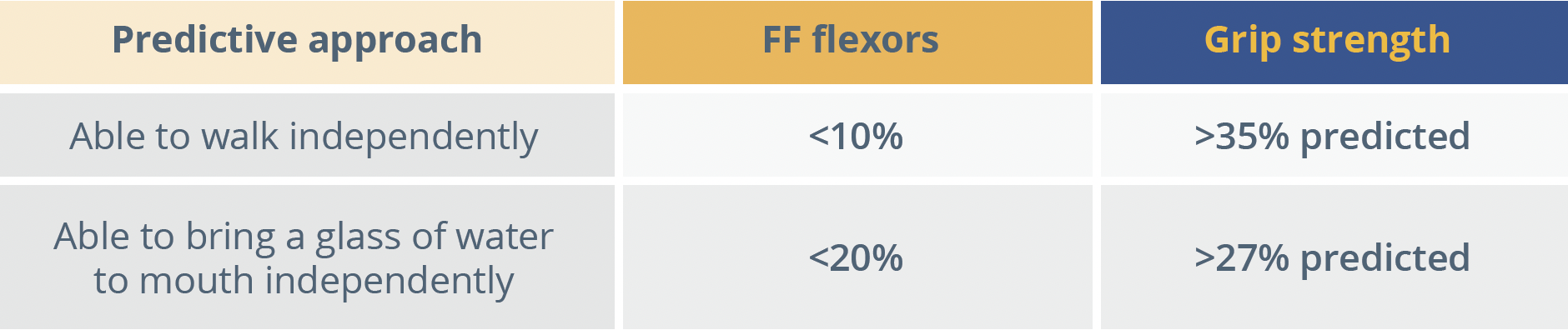
FF, fat fraction
“Clinically significant milestones for ambulatory and non-ambulatory DMD patients could be predicted by using a combination of strength and structure outcomes.”
- Charlotte Lilien, Oxford, UK
From bench to bedside in neuromuscular disorders
Generation of preclinical data in relevant models of neuromuscular disease is vital in steering the clinical development of new treatment options. Yuanfan Zhang, Boston, USA, presented research exploring the overexpression of Jagged1 as a potential therapeutic modifier for DMD. Genetic modifiers in DMD are gene variants or differences in protein expression with the potential to modify disease progression in patient or animal models. Jagged1 is a Notch ligand which has been shown to be upregulated in escaper golden retriever muscular dystrophy dogs (GRMDs) compared with affected animals.

Jagged1 expression in golden retrievers, mice and zebrafish.
At 1 month, mdxJAG1 mice showed less regeneration/regeneration than mdx and at 4 months showed more new regeneration. At 1 year, although the double transgenic mice had similar fibre size distribution to mdx, there was more robust regeneration in the mdxJAG1 muscle compared to mdx as shown by immunofluorescence and quantitative polymerase chain reaction (qPCR).
At 1 year, mdxJAG1 mice had bigger muscles and significantly smaller hearts, yet showed improved cardiac function. Left ventricular hypertrophy was mitigated by mdxJAG1 compared to mdx as measured by echo. Double transgenic mice also performed better in physiology and behavioural assays, showing improved force-frequency response and more activity in the open field behavioural test versus mdx. Imaging revealed a greater degree of fibrosis in the hearts and diaphragms of mdx versus mdxJAG1 mice. Overall, Dr Zhang concluded that mdxJAG1 mice benefited from both improved cardiac phenotypes and improved physiological and functional performance at 1 year compared with their counterpart mice - fueled by the consistent overexpression of Jagged1 in the muscle. These translational research findings highlight Jagged1 as a potential target for therapeutic intervention in DMD.
Progression of muscular dystrophies

Progression of muscular dystrophies
Esther Fernandez-Simon, Newcastle, UK, outlined research to unravel the molecular pathways involved in fibrosis progression in DMD, focusing on pathways triggered by platelet-derived growth factor (PDGF)-AA. The study methodology employed fibro-adipogenic progenitors (FAPs) from three patients with DMD who were treated with sequential doses of PDGF-AA. Proteomic analysis by mass spectrometry revealed that proteins of the RhoA/Rho-associated protein kinase-2 (ROCK2) pathway were upregulated in PDGF-AA-treated compared with non-treated human FAPs. This important pathway regulates cytoskeletal and cell adhesion dynamics and thereby coordinates a wide range of cellular processes including cell migration, cell polarity and cell cycle progression that are characteristic of the initial stages of fibrosis, Dr Fernandez-Simon explained.
In vitro testing revealed that the RhoA/ROCK2 pathway was increased in patients with DMD but activation by PDGF-AA could be inhibited by pretreatment with C3-exoenzyme or fasudil. Further in vitro analysis showed that inhibition of the RhoA/ROCK2 pathway reduced proliferation, migration and the amount of collagen released by DMD FAPs compared to PDGF-AA-activated cells.
Following on from these successful in vitro results, Dr Fernandez-Simon’s group proceeded to study fasudil treatment in a murine model of DMD. Treated mice were found to perform better on the grip strength test. Collagen I expression was also reduced and there was a tendency towards decreased expression of PDGF-alpha and macrophages in fasudil-treated animals. Overall, this research demonstrates that the RhoA/ROCK2 pathway is enhanced in DMD compared with healthy controls and that PDGF-AA activates this pathway - promoting proliferation, migration and action reorganisation which is essential for the development of fibrosis in muscular dystrophies. Inhibition of this pathway in vivo promoted an increase in force and a reduced expression of collagen in fasudil-treated mice, providing a potential avenue for therapeutic intervention, Dr Fernandez-Simon concluded.
“Our results support that the inhibition of RhoA/ROCK2 pathway could be considered a potential therapeutic target for muscle fibrosis in patients with muscular dystrophies.”
- Esther Fernandez-Simon, Newcastle, UK
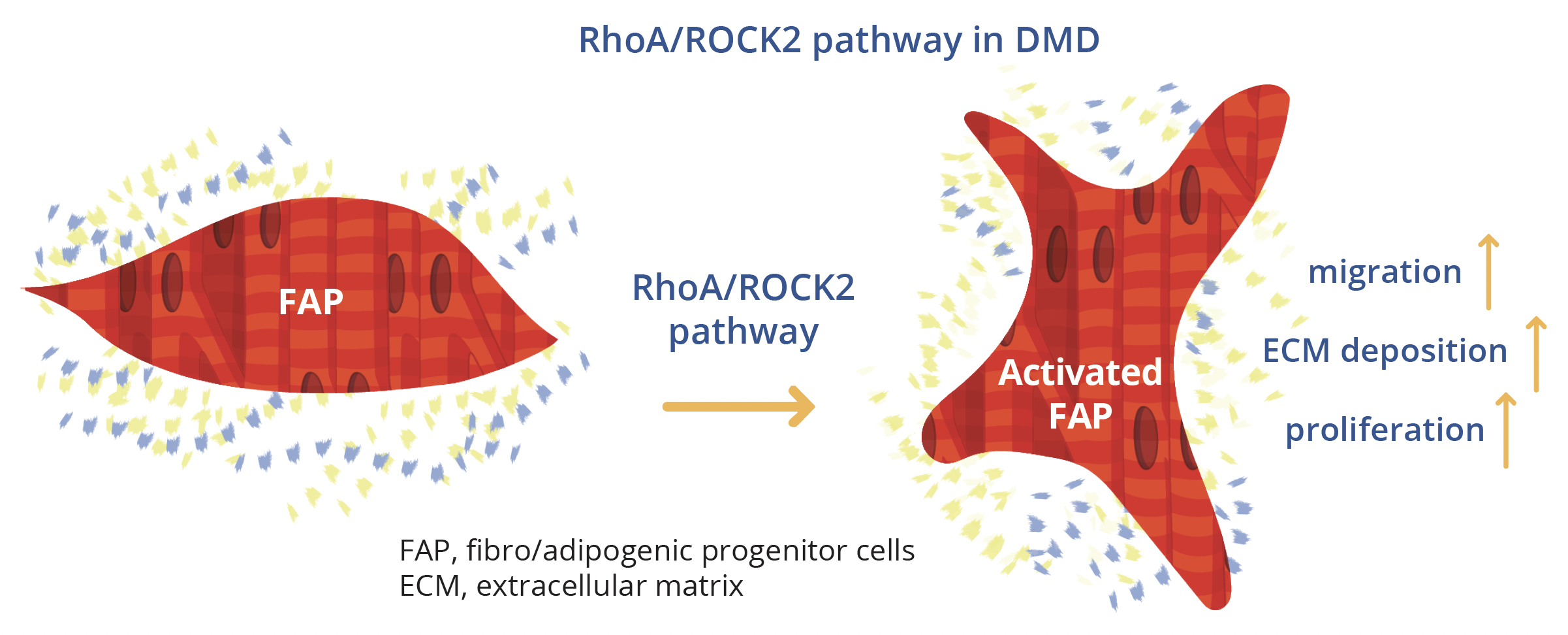
The RhoA/ROCK2 pathway in DMD compared with healthy controls
What’s new in neuromuscular research?
Considered one of the highlights of the annual WMS congress, this late breaking news session provided a platform to showcase previously unpublished high quality, high impact research findings in the field of neuromuscular diseases, pathobiological mechanisms and therapies.
Mariko Taniguchi-ikeda, Aichi, Japan, examined evidence suggesting that biallelic variants in LIG3 can cause a novel mitochondrial neurogastrointestinal encephalopathy (MNGIE). MNGIE is a rare, intractable mitochondrial disease resulting from a dysfunction of mitochondrial DNA (mtDNA) replication which leads to a decrease in mtDNA copy number and impaired adenosine triphosphate (ATP) production. Thymidine phosphorylase (TYMP) was the first gene linked to MNGIE to be discovered and 15 other causative genes have now been identified. All have functions involving the maintenance of mitochondrial nucleotide pools and mtDNA replication. Dr Taniguchi-ikeda’s team have recently described a novel mitochondrial syndrome caused by loss of function in the LIG3 gene. The LIG3 gene encodes for the only DNA ligase (DNA ligase III) found in mitochondria, although this is not essential for cell survival as DNA ligase I can compensate for nuclear DNA ligation. In total, six variants in LIG3 were detected across three independent families. Affected patients showed various phenotypes associated with MNGIE, characterised by chronic intestinal pseudo-obstruction (CIPO), myopathic changes and neurological impairment. The copy number of mtDNA was shown to be depleted in patient cells and gut peristalsis also proved abnormal in LIG3 knockout zebrafish. Addition of glutamine was effective in increasing mtDNA copy number, with mutant cells growing better in a glutamine-enriched medium. A ketogenic diet also proved effective in ameliorating epileptic discharges.
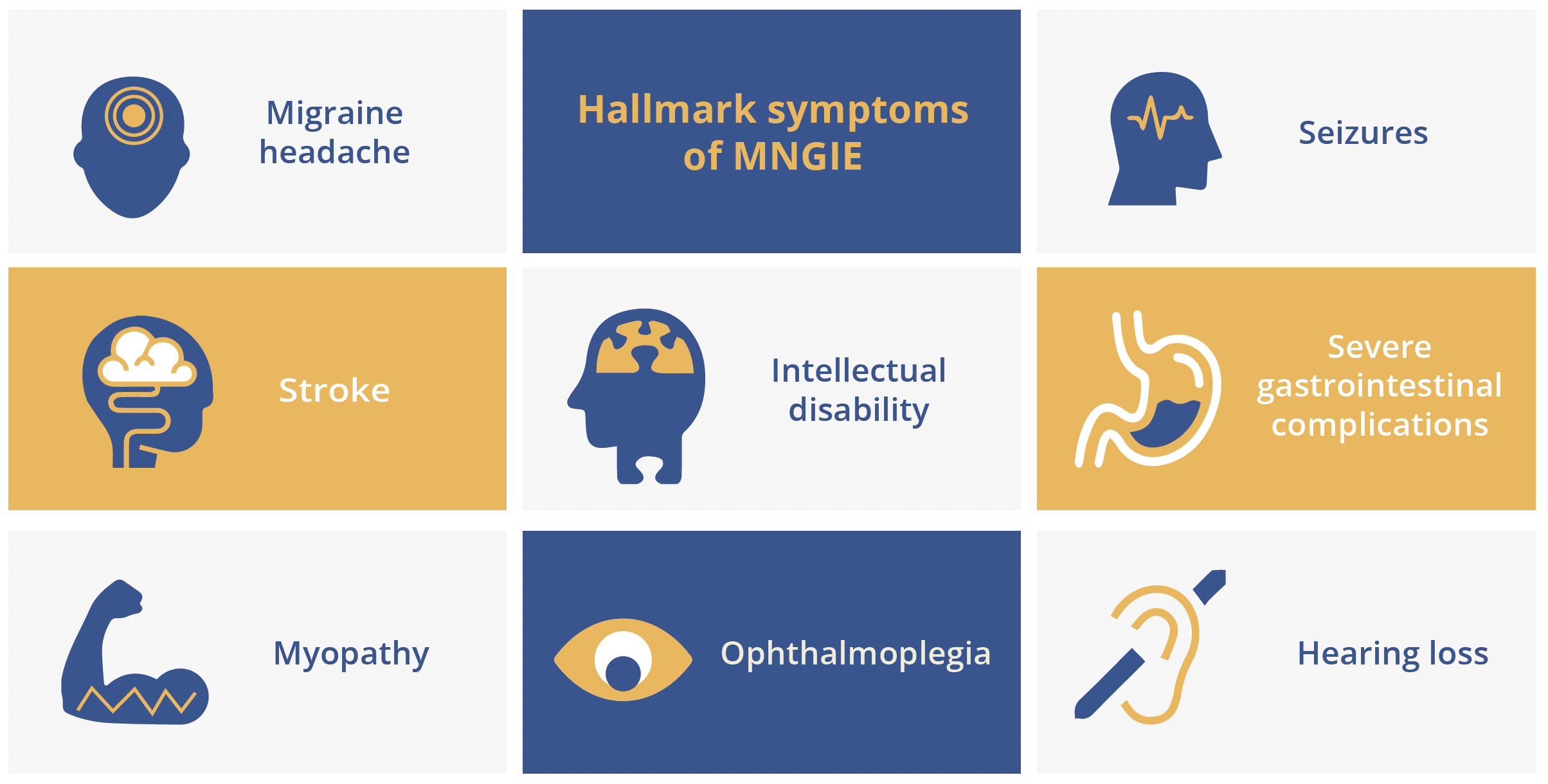
Hallmark symptoms of MNGIE
Macarena Cabrera-Serrano, Perth, Australia, presented research findings showing how biallelic loss-of-function variants in the OBSCN (obscurin, cytoskeletal calmodulin and titin-interacting RhoGEF) gene may predispose individuals to severe, recurrent rhabdomyolysis. This large, multicentre, collaborative project started with a single proband and culminated in the identification of six probands with rhabdomyolysis, harbouring a total of ten loss-of-function OBSCN variants. Patients presented with a range of different muscle pathologies, for example, abnormal glycogen accumulation and focal Z-band streaming. However, cardiac involvement was not noted in any proband or carrier relative. All variants were associated with near complete loss of OBSCN transcripts and proteins, with 8 out of 10 resulting in loss-of-function variants in isoforms A and B of obscurin.
OBSCN variants should therefore be considered in patients presenting with childhood/early adult onset severe and current rhabdomyolysis, concluded Dr Cabrera-Serrano. Further studies are needed to provide a comprehensive picture of the complex OBSCN splicing pattern and identify how loss of obscurin can trigger rhabdomyolysis.
“Given the large size of obscurin it is tempting to speculate that OBSCN variants may underlie a substantial proportion of unsolved rhabdomyolysis cases.”
- Macarena Cabrera-Serrano, Perth, Australia

Craig McDonald, Sacramento, USA, presented findings from the HOPE-2 multicentre, randomised, double-blind, placebo-controlled US clinical trial of intravenous human cardiosphere-derived cells (CDCs) for late-stage DMD (NCT03406780). CAP-1002 is a biologic consisting of allogeneic CDCs manufactured from donated human heart muscle. Dr McDonald explained that cells are infused intravenously and do not act by ‘stemness’ or engraftment into host tissue but rather home to the pulmonary vasculature where they secrete exosomes. These exosomes contain miRNA, non-coding RNAs and proteins which are immunomodulatory and have disease-ameliorating properties on dystrophic muscle. In preclinical animal models of dystrophinopathy, CAP-1002 has been shown to target multiple disease processes underpinning DMD.
CAP-1002 targets multiple disease processes in DMD
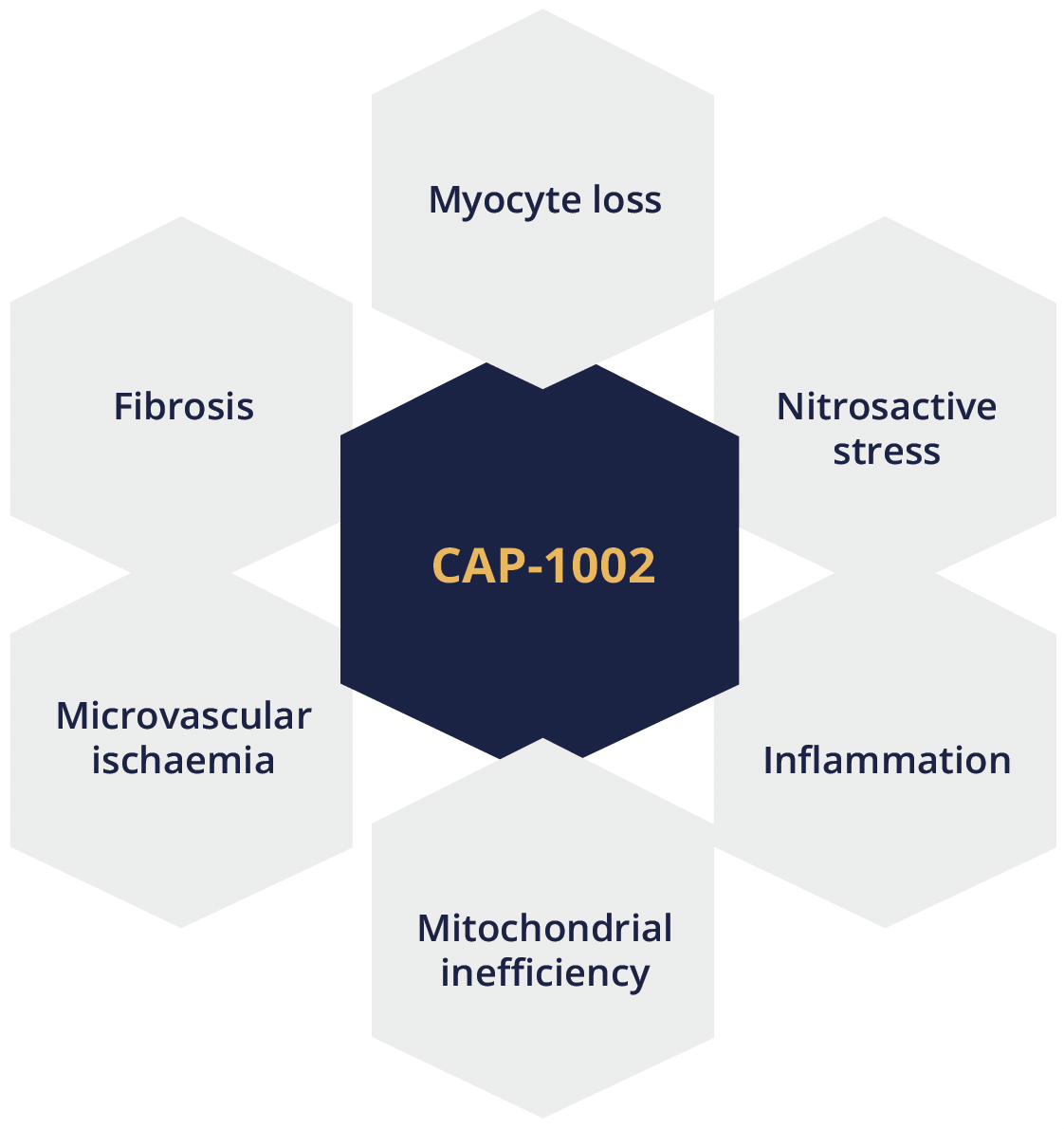
CAP-1002 is a biologic consisting of allogeneic CDCs manufactured from donated human heart muscle and has been shown to target multiple disease processes underpinning DMD
To date, CAP-1002 has been investigated in multiple independent clinical trials involving over 200 people. The target population for the phase II HOPE-2 study clinical trial was late ambulatory patients with impaired upper limb function and non-ambulatory patients with impaired overhead reach but retained hand-to-mouth function. Inclusion criteria included a PUL entry item score from 2–5. Patients’ mean age was 14.3 years, all patients were on corticosteroids and ~80% were ambulatory. HOPE-2 was one of first trials in DMD to use both the performance of upper limb (PUL 1.2) and the recently validated PUL 2.0 as clinical endpoints, noted Dr McDonald. Patients (n=20) were treated with 150 million cells every 3 months and the primary endpoint (mid-level PUL1.2) was evaluated after 12 months.
Overall, a total of 69 infusions of CAP-1002 or placebo were performed in HOPE-2 and the product proved generally well-tolerated. With the exception of hypersensitivity reactions (which were ameliorated by pretreatment with high-dose steroids and histamine-2 [H2] blockers), no safety signals were identified. The 12-month results for the final efficacy analysis showed significant preservation of the mid-level PUL 1.2 in CAP-1002-treated patients relative to placebo, with 71% relative slowing (p=0.01). Function was improved by 2.6 points. Similarly, there was a statistically significant preservation of the total PUL 1.2, with 70% relative slowing (p=0.02) and a 3.2 point relative improvement in treated versus placebo patients. Both total PUL 1.2 and total PUL 2.0 showed statistically significant treatment effects, with all PUL dimensions responsive to CAP-1002 with the exception of distal items which were well-preserved in both groups.
HOPE 2: Primary endpoint
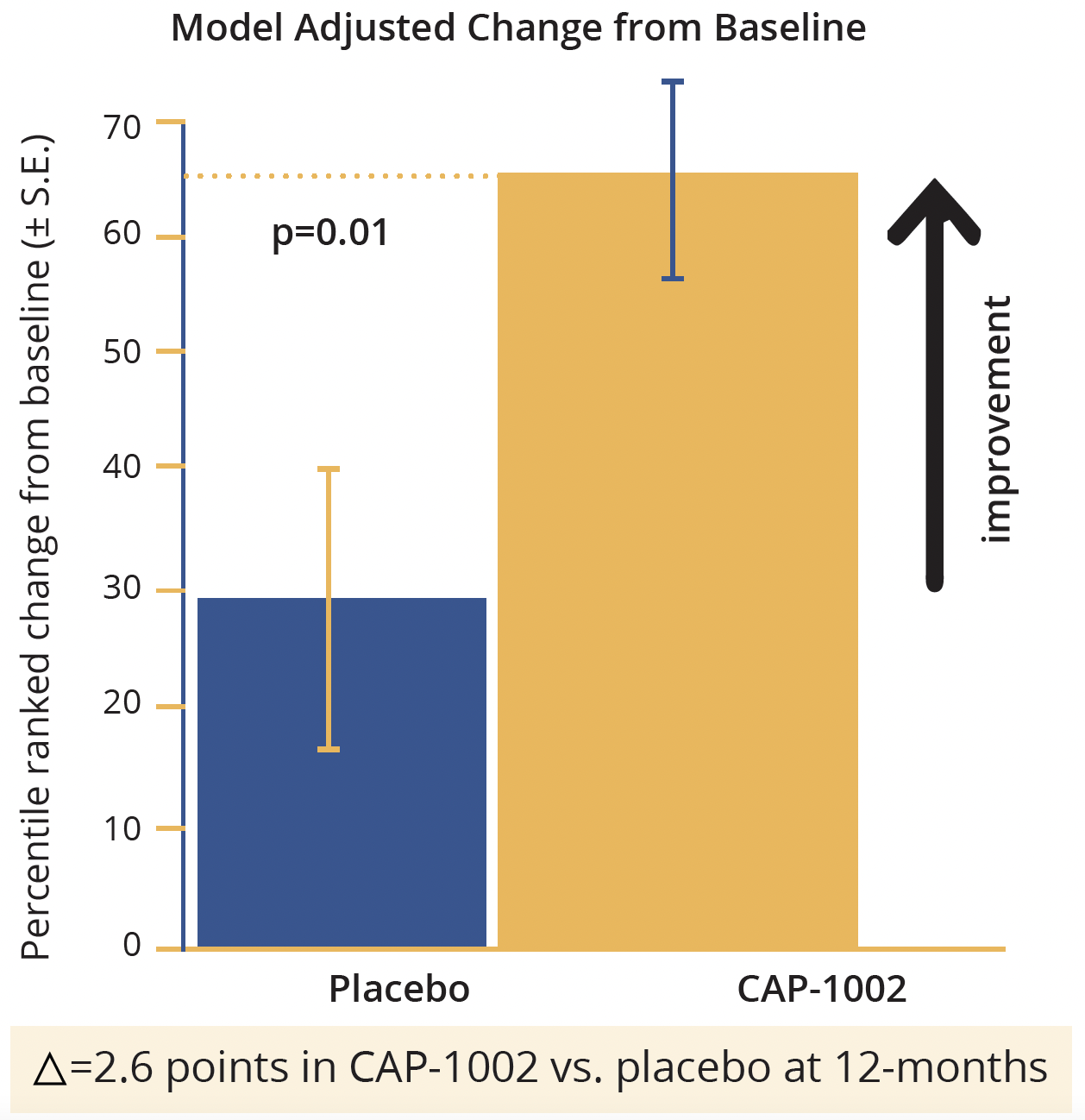
Model adjusted change from baseline in points in CAP-1002 in the HOPE-2 study
Left ventricular ejection fraction (LVEF) by cardiac MRI showed 107% relative slowing with a preservation of LVEF of 4% relative to placebo (p=0.002), which Dr McDonald described as ‘impressive’. Measures of both cardiac structure and function also showed a positive response to CAP-1002. The ratio of creatine kinase MB to total creatine kinase, which is a validated biomarker for cardiac injury and showed a significant reduction in CAP-1002-treated patients (p=0.02).
Based on these findings from HOPE-2, Dr McDonald concluded that CAP-1002 appears to be well-tolerated and effective in attenuating the deterioration of upper limb and cardiac function in late-stage DMD. Current ongoing extension studies may confirm therapeutic durability and safety beyond 12 months in non-ambulant DMD. HOPE-3, the pivotal phase III study of CAP-1002, is currently in the planning stages.
“A therapy that stabilises or reverses cardiac deterioration while improving upper limb function would be unique in its ability to address, synergistically, the tremendous burden of disease seen in non-ambulatory DMD patients.”
- Craig McDonald, Sacremento, USA
Closing Remarks
The final session of this year’s WMS congress, which was attended by a total of around 1200 registered delegates, consisted of prize presentations and a closing address by President Straub. In an established tradition of the society, Dr Straub performed the ceremonial handing over of the flag to the Co-chairs of the 2022 meeting Jiri Vajsar, Toronto, Canada, and Jim Dowling, Toronto, Canada. The WMS is ‘focusing all of its energy’ on delivering an in person, face-to-face congress next year, which is planned to take place between the 11th and 15th October 2022 in Halifax, Nova Scotia. For what will be the first ever WMS congress hosted in Canada, we aim to provide ‘an outstanding meeting with a wonderful group of speakers’, promised Dr Dowling.
©Springer Healthcare 2021. This content has been independently selected and developed by Springer Healthcare and licensed by Roche for Medically. The topics covered are based on therapeutic areas specified by Roche. This content is not intended for use by healthcare professionals in the UK, US or Australia. Inclusion or exclusion of any product does not imply its use is either advocated or rejected. Use of trade names is for product identification only and does not imply endorsement. Opinions expressed do not reflect the views of Springer Healthcare. Springer Healthcare assumes no responsibility for any injury or damage to persons or property arising out of, or related to, any use of the material or to any errors or omissions. Please consult the latest prescribing information from the manufacturer for any products mentioned in this material.