
The 2020 annual International Congress of the European Respiratory Society (ERS) was held as an interactive virtual event. The ERS used the latest technology to join together over 33,000 delegates from around the world. In the midst of the current COVID-19 pandemic, the ERS were determined that the congress should still take place as it could be more important than ever to connect the respiratory community, share data and to provide educational opportunities.


An overview of sessions and attendees at ERS 2020
Welcome to ERS 2020
The President of ERS, Thierry Troosters, Leuven, Belgium, hoped that the digital format of the congress would provide an opportunity for people from all around the globe to connect in large numbers. He anticipated it would provide fertile ground to exchange ideas to move respiratory health forward into the next decade. Hot topics include the relationship between exercise, physical activity and sleep quality and the increasing role of digital interfaces to support patients in between face-to-face appointments.
Interstitial lung disease
Idiopathic pulmonary fibrosis (IPF) is an interstitial lung disease (ILD) characterised by chronic inflammation, which is accompanied by an uncontrolled healing response that causes fibrosis of the alveolar tissue.
Athol Wells, London, UK, spoke about the role of computed tomography (CT) in the recently-revised diagnostic criteria for IPF by joint societies and explained the differences compared with the Fleischner Society White Paper published in the same year. A high-resolution CT scan is central to the diagnostic process in a patient with newly-detected ILD. In some patients, CT can reveal classical usual interstitial pneumonia (UIP) of predominant subpleural basal disease with honeycombing, leading to an immediate diagnosis of UIP. In other patients, the CT scan reveals a pattern of ‘probable UIP’ or ‘indeterminate for UIP’, and further testing is required. The recommendations are generally comparable but the joint guidelines recommend performing a surgical lung biopsy in probable UIP since they aim to make a definite diagnosis, whereas the Fleischner statement leads to a working diagnosis of IPF and initiation of therapy. In real-world practice, most clinicians follow the Fleischner recommendation and initiate therapy in patients with probable UIP.
“What exactly are we trying to achieve in the diagnosis of IPF: certain diagnosis or reasonable management?”
- Athol Wells, London, UK

The application of joint guidelines versus Fleischner statement in diagnosing IPF
The development of antifibrotic drugs, such as pirfenidone and nintedanib, has changed the treatment landscape of patients with IPF, improving morbidity and mortality compared with previous use of immunosuppressants. However, Sara Tomassetti, Florence, Italy, explained that there are limitations of these new treatments so there is still a requirement for lung transplantation in patients with interstitial diseases. Antifibrotics represent a bridge to lung transplant but not an alternative. Therefore, the clinical challenge is to recognise the optimal time window to refer a patient for transplant. To help with this decision, studies are investigating new scoring systems, novel biomarkers and the use of AI to stratify patients according to prognosis. Importantly, data show that the use of antifibrotic drugs does not increase surgical complications or post-operative mortality after transplant so they can be used to attenuate disease progression while awaiting lung transplant.

Antifibrotics represent a bridge to lung transplant but not an alternative
Jens Gottlieb, Hannover, Germany, presented some facts and figures of transplants. Registry data of 69,000 adult lung transplants shows that patients with CF were the youngest and the proportion of transplants attributed to CF has hardly changed since the 1990s. Conversely, the proportion of patients with IPF has increased. The median age of all lung transplant recipients is steadily increasing and is approaching 60 years. Survival at 1 year is highest among patients with CF at 85%, and lowest in IPF at 80%. The main cause of death within one year of lung transplant is infection or failure of graft. After a year it is chronic lung allograft dysfunction, which encompasses a range of pathologies that cause a transplanted lung to not achieve or maintain normal function and this occurs in approximately a third of patients according to the new definition from the International Society for Heart and Lung Transplantation (ISHLT). Many retrospective analyses have shown survival benefit of transplantation. Dr Gottlieb concluded that quality of life is improved by lung transplant in most patients with advanced lung disease.
The typical experience of patients with IPF is of an illness that appears suddenly, without warning, and patients and their caregivers have difficultly recognising the magnitude of the disease. Although disease progression varies between patients, life expectancy is low with a median survival of 3.8 years. Patients with IPF have limited treatment options, rely on supplemental oxygen and caregivers and will ultimately need palliative care. At what point should palliative care be offered? Kathleen Lindell, Pittsburgh, USA, explained that palliative care should be delivered alongside disease-directed therapies in serious illness and is therefore appropriate at any point. Dr Lindell and colleagues are investigating whether the early introduction of palliative care in the course of IPF through the SUPPORT™ programme could be helpful in terms of stress, symptom burden, poor quality of life, and poor care planning in patients with IPF and their caregivers. Preliminary data show that the intervention is feasible and acceptable to patients and their caregivers. Results from the analysis to determine how the programme impacts on the patients and their caregivers are expected soon.

The introduction of palliative care in the course of IPF
The latest techniques to study IPF were discussed in an educational symposium entitled ‘Human Ex Vivo Models of Interstitial Lung Disease’. The focus of the session was models of early events of IPF ex vivo and their use in evaluating pathogenesis and treatment
The first talk was given by Melanie Konigshoff, Denver, USA, who presented an ex vivo lung model using precision cut-lung slices from human tissue. The technique uses 300–500 micron slices of lung tissue, which are embedded hydrogels to improve viability and increase culture time. The tissue culture is exposed to a fibrotic cocktail of growth factors and cytokines to replicate IPF. The model has been validated using the FDA-approved treatments, pirfenidone and nintedanib, and offers a means to test novel drugs.
Killian Hurley, Dublin, Ireland, spoke about the use of induced pluripotent stem cells (iPSC) to study type II alveolar epithelial cells (ATII). A major challenge with iPSC models is their heterogeneity which means that a proportion of cells lose their phenotype during replication. To counteract this, the group have used a computation-based approach to single-cell RNA sequencing (scRNA-seq). A continuous branching network model learns, predicts and maps cell fate paths, including the precise timing of Wnt modulation, to allow the controlled differentiation of target cells with desired phenotypes. This emerging technology allows for indefinite expansion of induced ATII cells in culture. The technology has wider implications for improving differentiation protocols and providing resources for modelling human disease.
Microphysiological systems (MPS) are biometric devices recapitulating natural physiology of human or animal tissues in a microenvironment that induces in situ cell phenotype; organs-on-chip are an example. Nuria Roldan, Bern, Switzerland, explained that an organ-on-chip must take into account engineered aspects, such as air flow in the case of a lung model and biological aspects, for example correct composition of the extracellular matrix and the growth medium. Several lung-on-chip models are commercially available. Chips have been developed to simulate different physiological scenarios including immune cell recruitment during lung inflammation and thrombus formation, and can also simulate microvasculature. Further studies are required to validate the models to demonstrate that they are truly predictive for drug development and precision medicine.
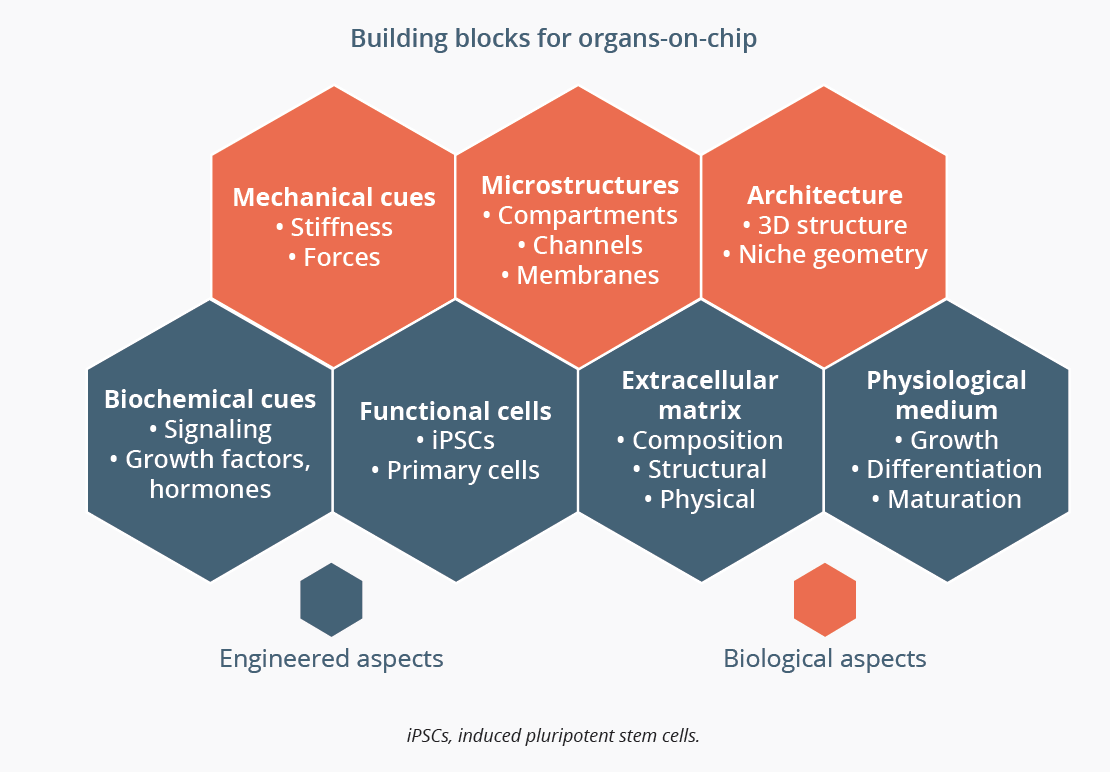
An organ-on-chip must take into account engineered and biological aspects
In contrast to the novel lung-specific models of lung disease that had been discussed previously in the session, Jennifer Dickens, Cambridge, UK, considered if there is still a role for traditional immortalised cell lines in ILD research. Mutations in surfactant protein C (SFTPC) are expressed in alveolar epithelial type II (ATII) cells and are known to cause familial ILD. The most common mutation causes alterations in SFTPC trafficking in ATII cells. Cell lines allow us to ask generic biological questions such as how the expression of a mutant SFTPC results in mistrafficking and cause ATII dysfunction. Although immortal cell lines are not ATII-like, they have the advantages of being easily manipulated, abundantly available and have existing reagents. They will help to identify novel drug targets and perform large scale initial screens before testing in more complex models.
“They will enable us to ask the right questions in more difficult to work with (but more physiological) systems.”
- Jennifer Dickens, Cambridge, UK
IPF and COVID-19
Latest techniques in IPF research were discussed in another educational symposium entitled ‘Hot Topics: (Re-)discovering the Small Airway Epithelium in COVID-19 and Fibrosing Lung Disease and Pulmonary Regeneration’. The focus of the session was small airway cells and their role in fibrosis and lung regeneration, and novel models to explore their potential as a therapeutic target.
Airway epithelium repair comprises of cell migration, proliferation and differentiation. In the normal repair process, airway basal cells (ABCs) are the stem cells of the airway epithelium, giving rise to ciliated and secretory cells. However, a variety of mechanisms and substances may impair or otherwise dysregulate repair, including infection, inflammation, inhaled toxicants and drugs. Many studies found that in areas of lung tissue damage in IPF, ATII cells have been replaced by ABCs. To investigate the role of these stem cells in fibrosis further, Antje Prasse, Hannover, Germany, isolated human ABCs and analysed their genetic material by scRNA-seq. Dr Prasse reported that there is a difference in genes found in ABCs from patients with IPF compared with those from non-fibrotic controls. Functional experiments went on to demonstrate crosstalk between ABCs and fibroblasts that promotes fibrosis in IPF and drives aberrant recapitulation of developmental processes.
“We found over a thousand genes that were differentially expressed by basal cells in IPF.” ”
- Antje Prasse, Hannover, Germany
Herbert Schiller, Munich, Germany, used scRNA-seq to model the differentiation processes involved in distal lung regeneration. By taking longitudinal cell samples from a murine model of lung injury they identified a novel transitional stem cell state named Krt8+ during the differentiation process of both airway and alveolar stem cells into type I pulmonary alveolar (ATI) cells. Similar transitional cells exist in human IPF and it was suggested that they may actively promote the disease through cell-cell communication.
“Potentially these basaloid cells represent a failed attempt at alveolar regeneration.”
- Herbert Schiller, Munich, Germany
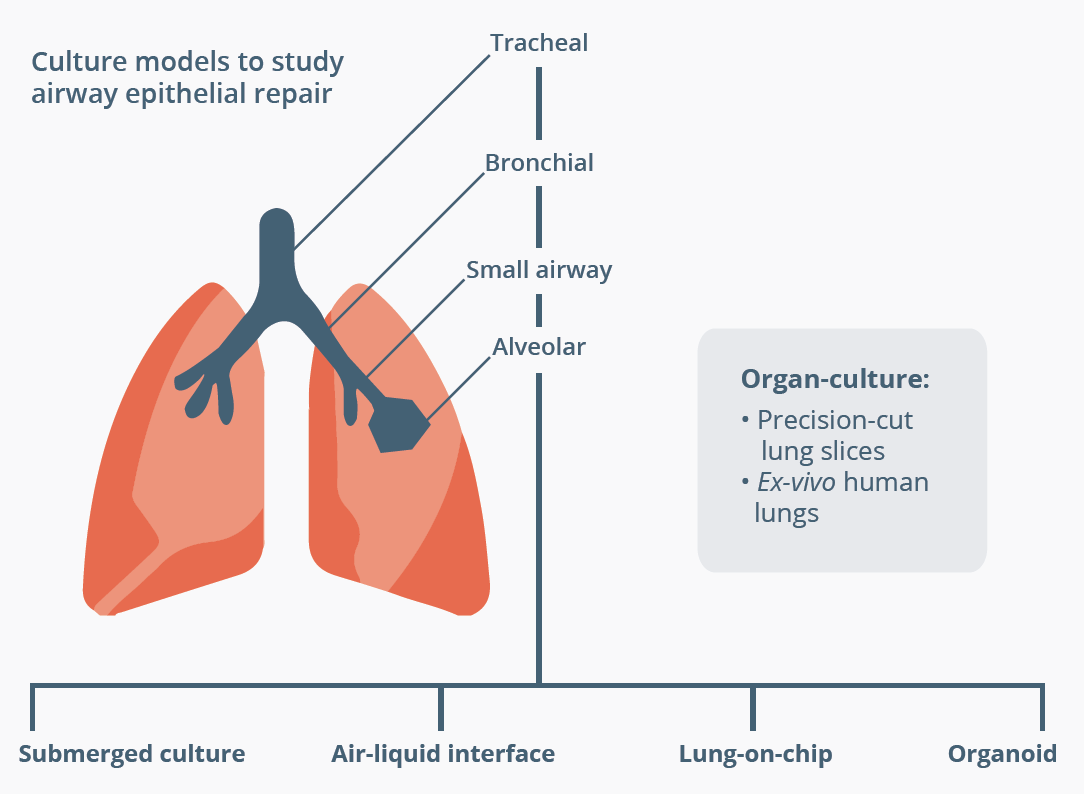
Pieter Hiemstra, Leiden, Netherlands, discussed how the refinement of many different techniques has led to an improvement in our understanding of human airway epithelial repair. Recent years have seen advances in culture models, cell sources and analysis. In vitro studies use either primary airway cells isolated from healthy controls or patients, or airway cells that have been derived from differentiated human induced pluripotent stem cells (hiPSC). Cell studies allow us to carry out functional studies in different populations. For example, data show decreased proliferation and wound-healing of cultured bronchial epithelial cells from children with preschool wheeze compared with cells from children with severe asthma or controls. Culture models that allow us to study multicellular tissue samples have progressed from submerged cultures to more complex models, such as air-liquid interface and lung-on-chip. For example, an air-liquid interface culture model demonstrated the positive effect of monocyte-derived macrophages on wound closure. These techniques provide novel information on human airway epithelial repair and may identify potential targets for treatment.
Closing Remarks
The next ERS International Congress will be held in Barcelona, Spain from 4–8 September 2021.
©Springer Healthcare 2020. This content has been independently selected and developed by Springer Healthcare and licensed by Roche for Medically. The topics covered are based on therapeutic areas specified by Roche. Inclusion or exclusion of any product does not imply its use is either advocated or rejected. Use of trade names is for product identification only and does not imply endorsement. Opinions expressed do not reflect the views of Springer Healthcare. Springer Healthcare assumes no responsibility for any injury or damage to persons or property arising out of, or related to, any use of the material or to any errors or omissions. Please consult the latest prescribing information from the manufacturer for any products mentioned in this material.